Deposition Date
2025-11-18
Release Date
2026-01-07
Last Version Date
2026-02-04
Entry Detail
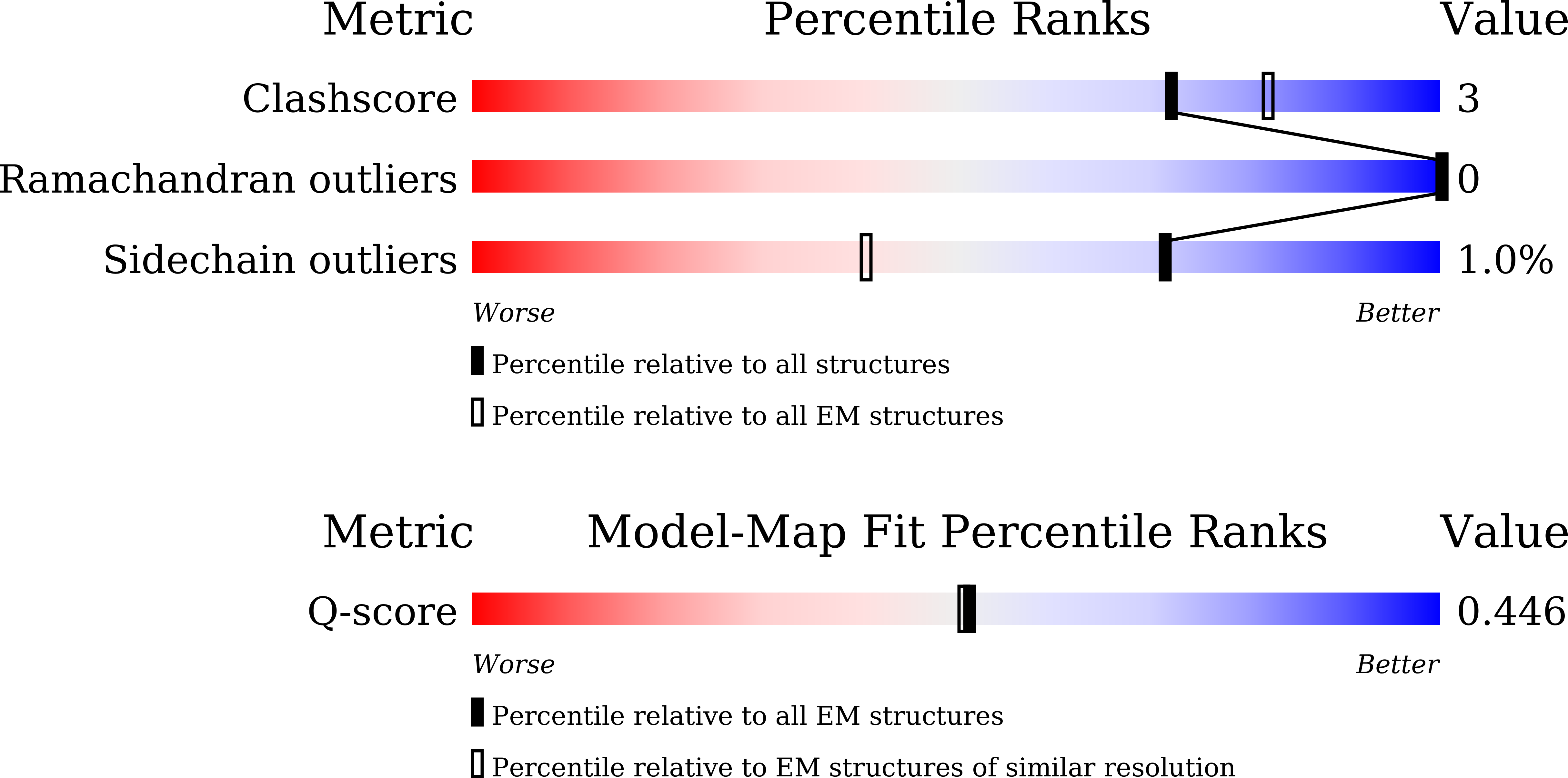
PDB ID:
9XQB
Keywords:
Title:
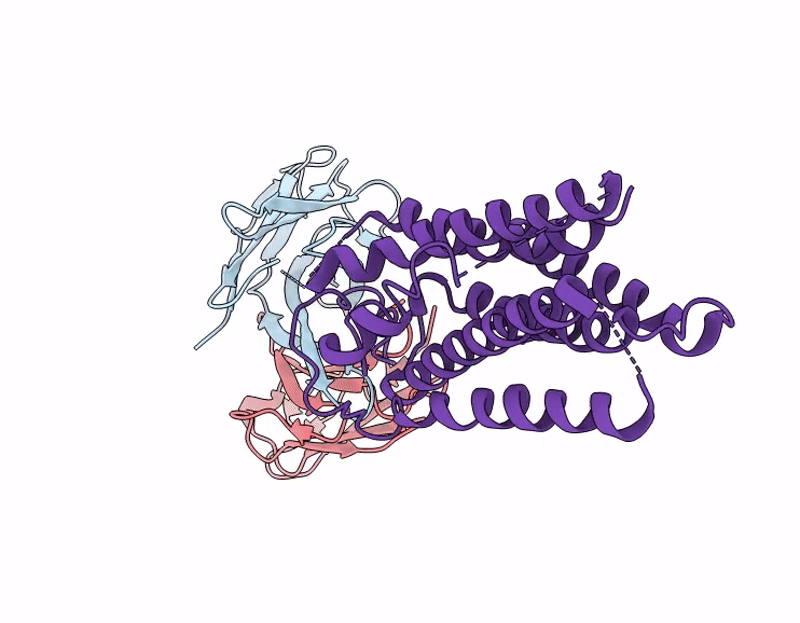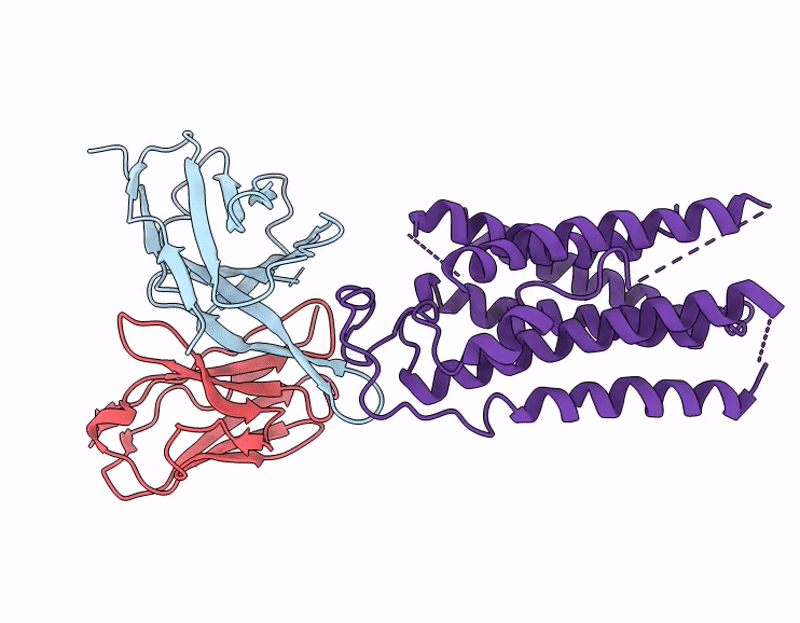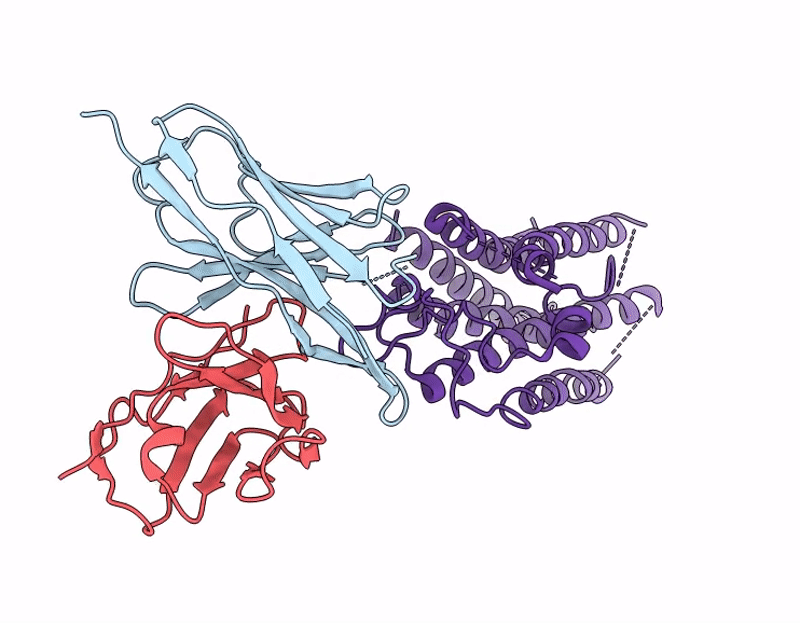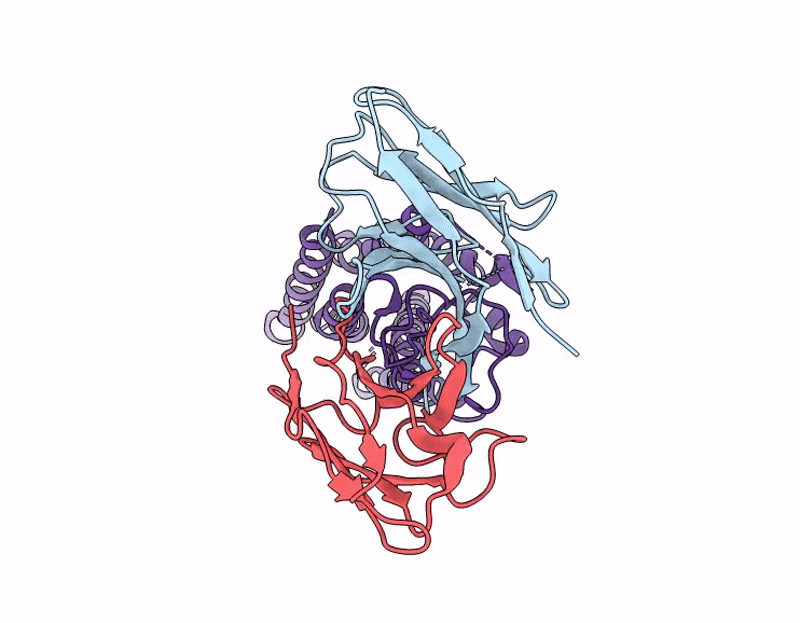
Cryo-EM structure of the human A2A adenosine receptor in complex with a Fab antibody fragment
Biological Source:
Source Organism(s):
Mus musculus (Taxon ID: 10090)
Homo sapiens (Taxon ID: 9606)
Homo sapiens (Taxon ID: 9606)
Expression System(s):
Method Details:
Experimental Method:
Resolution:
3.45 Å
Aggregation State:
PARTICLE
Reconstruction Method:
SINGLE PARTICLE