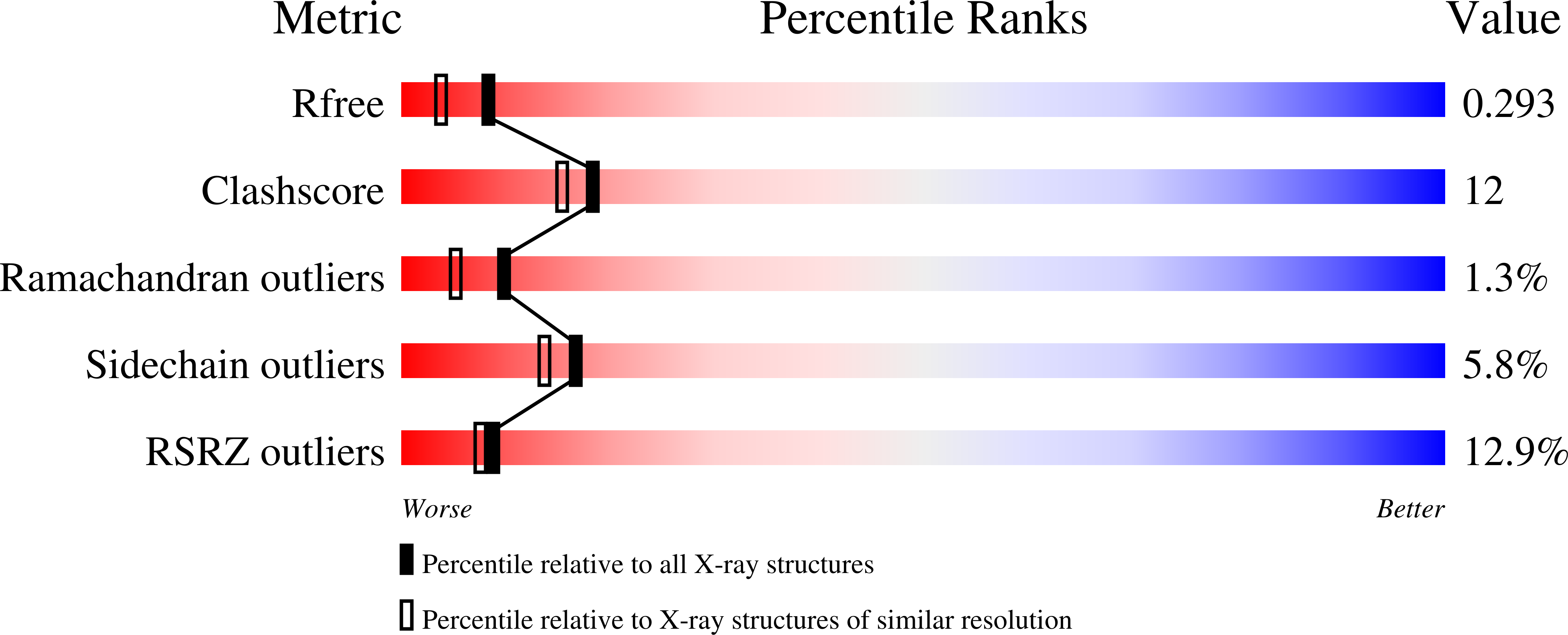
Deposition Date
2025-06-18
Release Date
2025-12-24
Last Version Date
2025-12-24
Entry Detail
PDB ID:
9P6C
Keywords:
Title:
RTX domain block V of adenylate cyclase toxin with mutations D1533N, A1542N, D1560N, S1569N, D1587N, H1598N, H1608N
Biological Source:
Source Organism(s):
Bordetella pertussis (Taxon ID: 520)
Expression System(s):
Method Details:
Experimental Method:
Resolution:
1.99 Å
R-Value Free:
0.29
R-Value Work:
0.22
R-Value Observed:
0.23
Space Group:
P 1