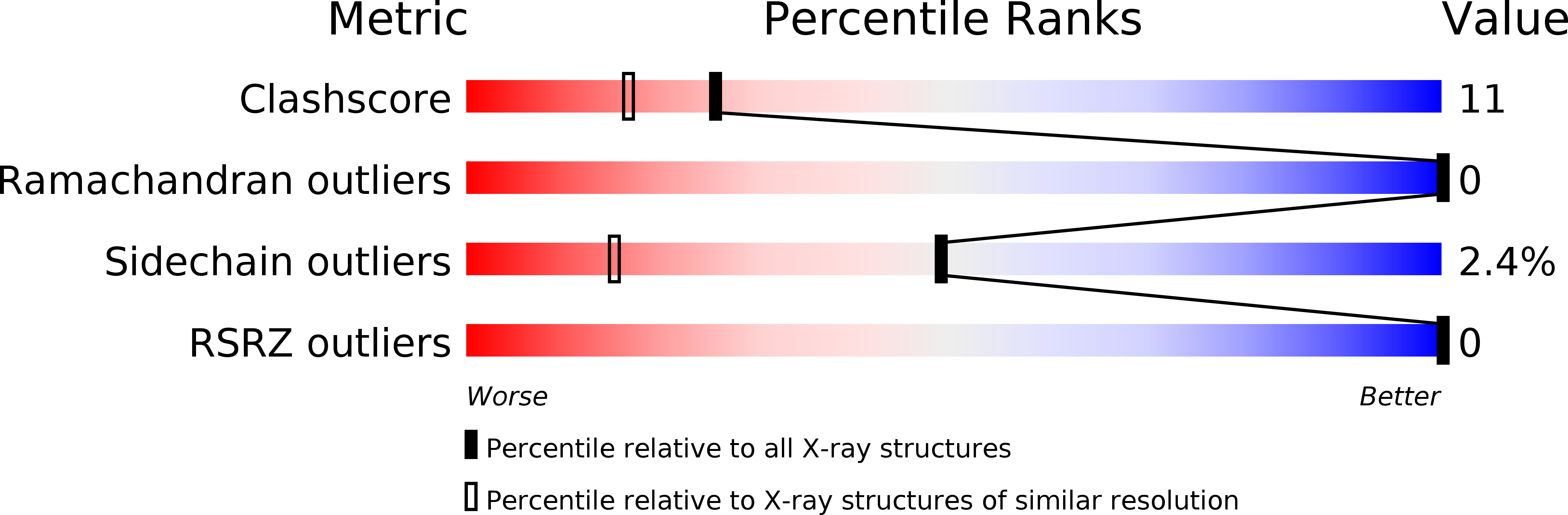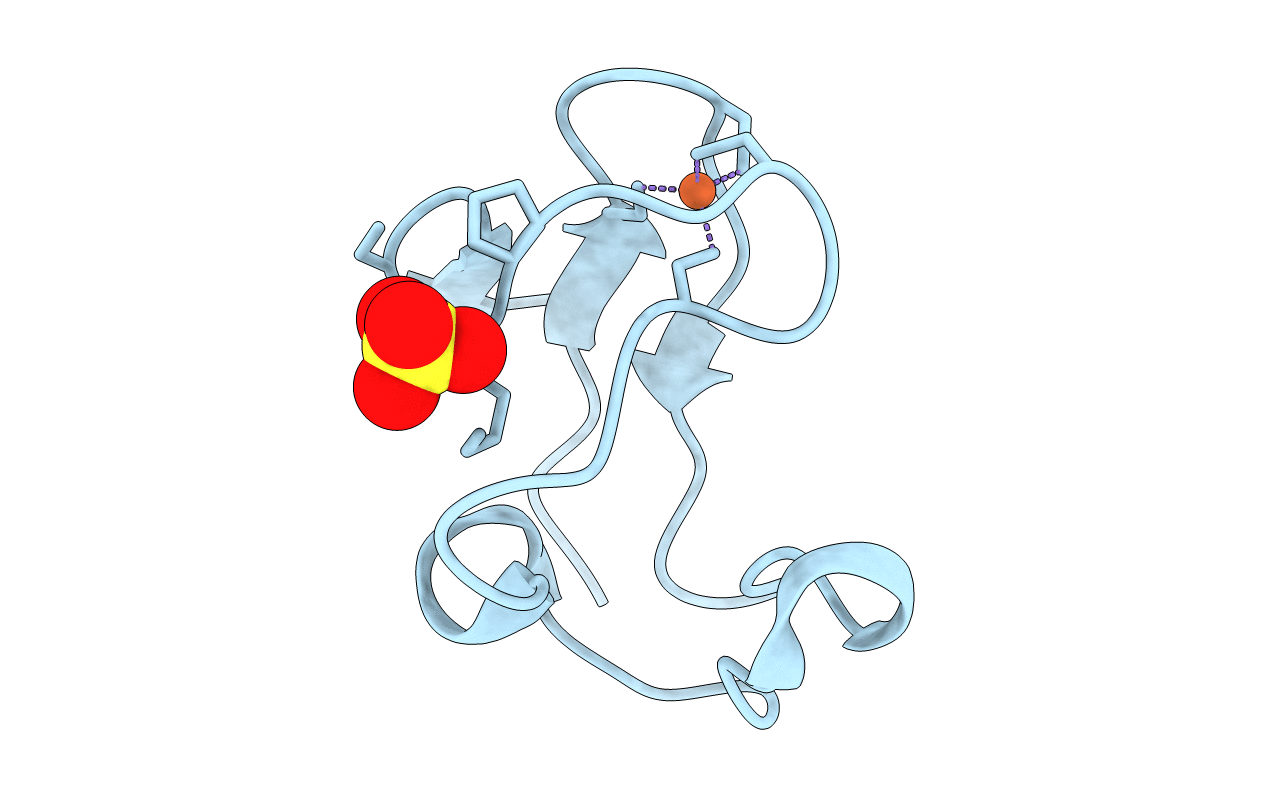
Deposition Date
1991-08-26
Release Date
1993-10-31
Last Version Date
2024-02-14
Entry Detail
PDB ID:
8RXN
Keywords:
Title:
REFINEMENT OF RUBREDOXIN FROM DESULFOVIBRIO VULGARIS AT 1.0 ANGSTROMS WITH AND WITHOUT RESTRAINTS
Biological Source:
Source Organism:
Desulfovibrio vulgaris (Taxon ID: 881)
Method Details:
Experimental Method:
Resolution:
1.00 Å
R-Value Observed:
0.14
Space Group:
P 1 21 1