Deposition Date
2022-12-23
Release Date
2023-04-12
Last Version Date
2025-07-23
Entry Detail
PDB ID:
8C3E
Keywords:
Title:
Engineered mini-protein LCB2 (blocking ligand of SARS-Cov-2 spike protein)
Biological Source:
Source Organism(s):
synthetic construct (Taxon ID: 32630)
Expression System(s):
Method Details:
Experimental Method:
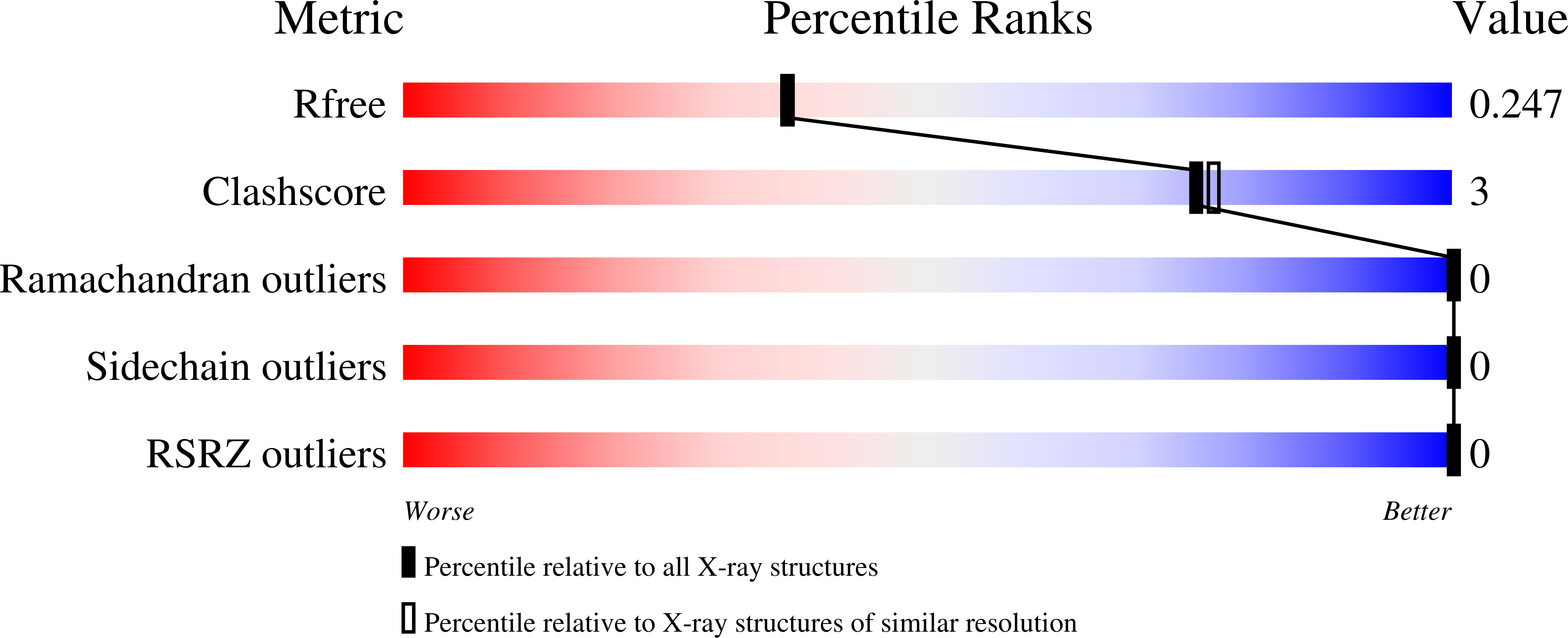
Resolution:
2.10 Å
R-Value Free:
0.24
R-Value Work:
0.20
R-Value Observed:
0.22
Space Group:
P 31 2 1