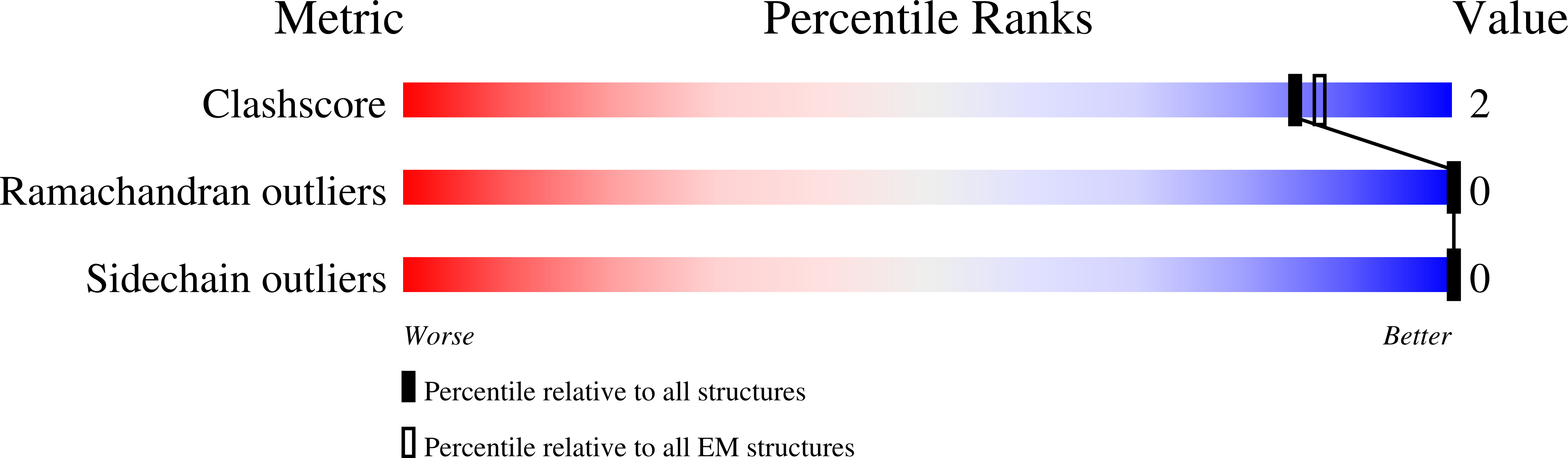
Deposition Date
2022-04-07
Release Date
2022-07-27
Last Version Date
2024-03-27
Entry Detail
PDB ID:
7UMQ
Keywords:
Title:
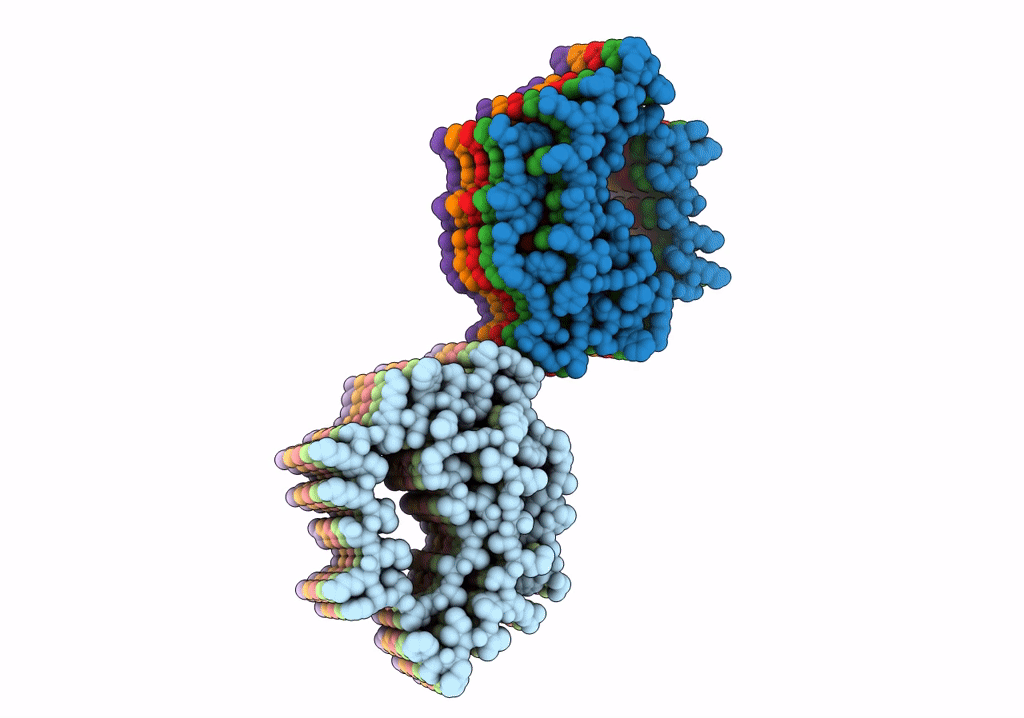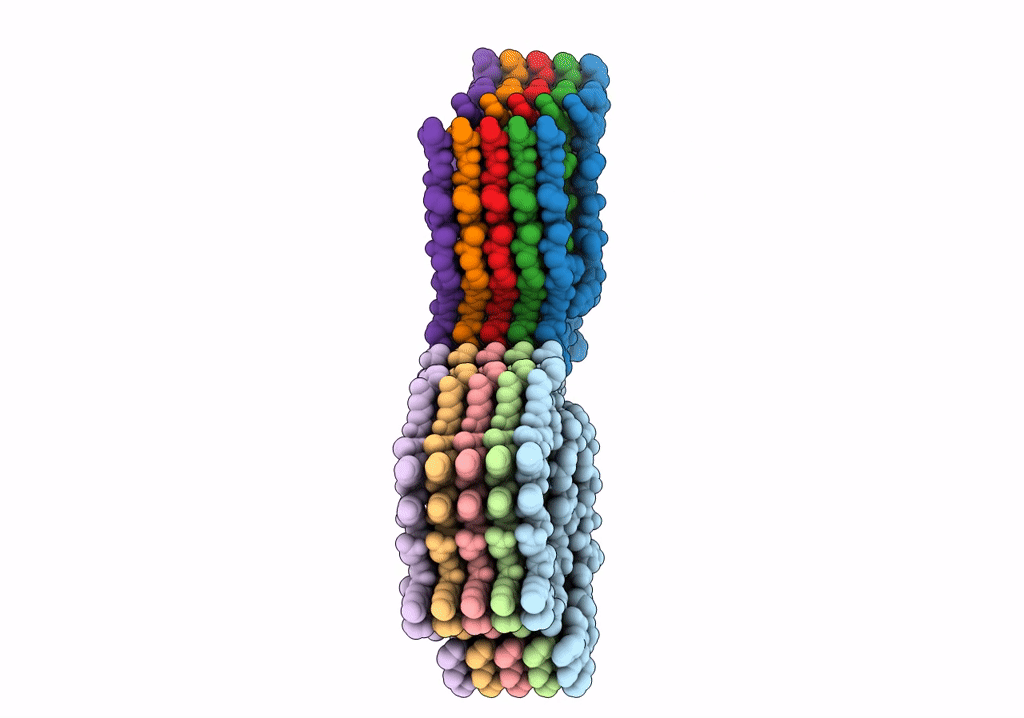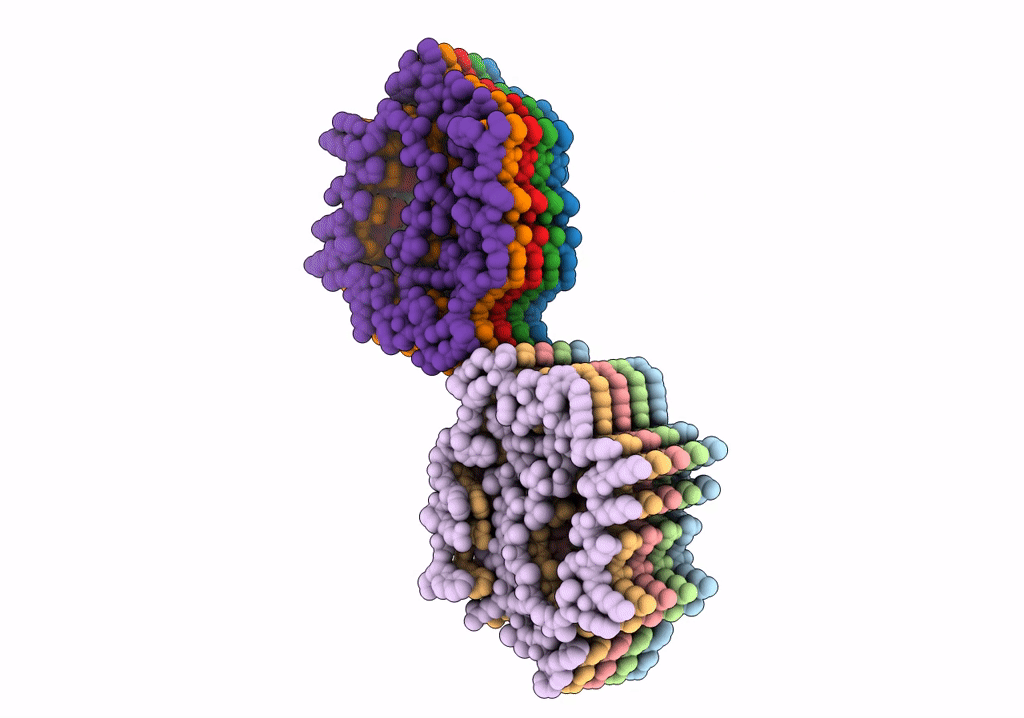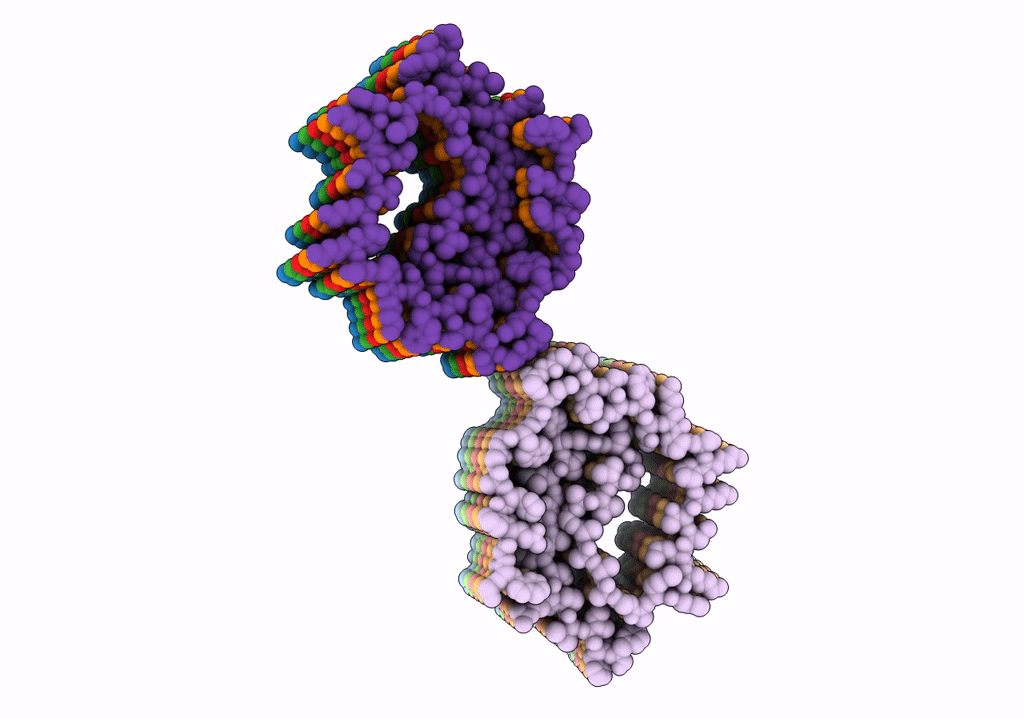
Structure of Type I Prion filaments from Gerstmann-Straussler-Scheinker disease
Biological Source:
Source Organism(s):
Homo sapiens (Taxon ID: 9606)
Method Details:
Experimental Method:
Resolution:
3.29 Å
Aggregation State:
FILAMENT
Reconstruction Method:
HELICAL