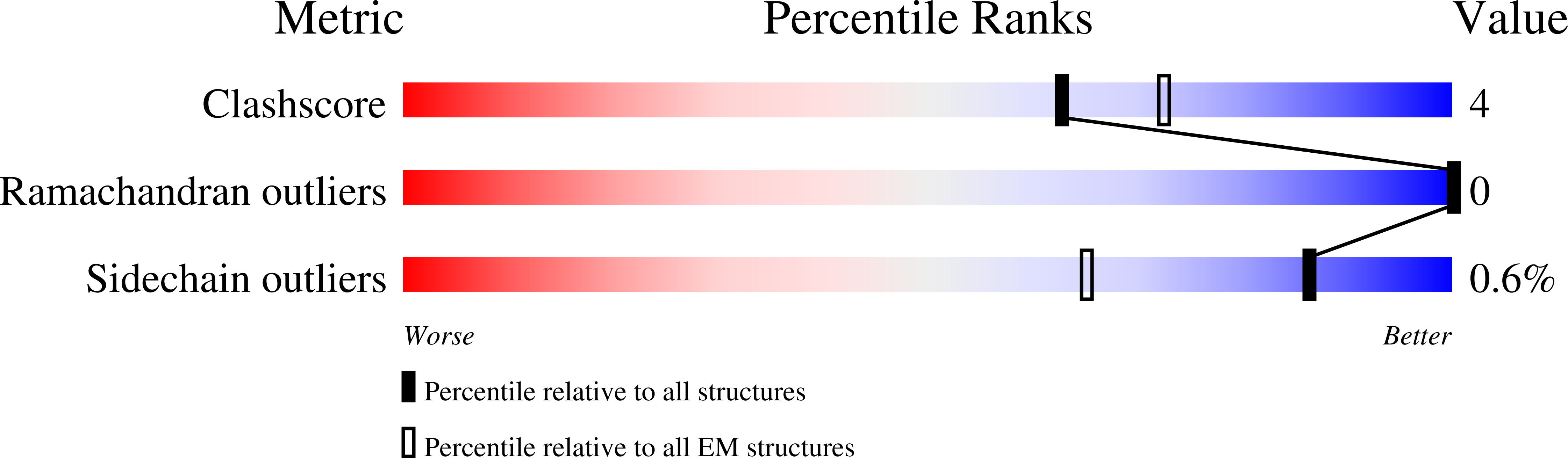
Deposition Date
2021-11-04
Release Date
2022-02-02
Last Version Date
2023-12-13
Entry Detail
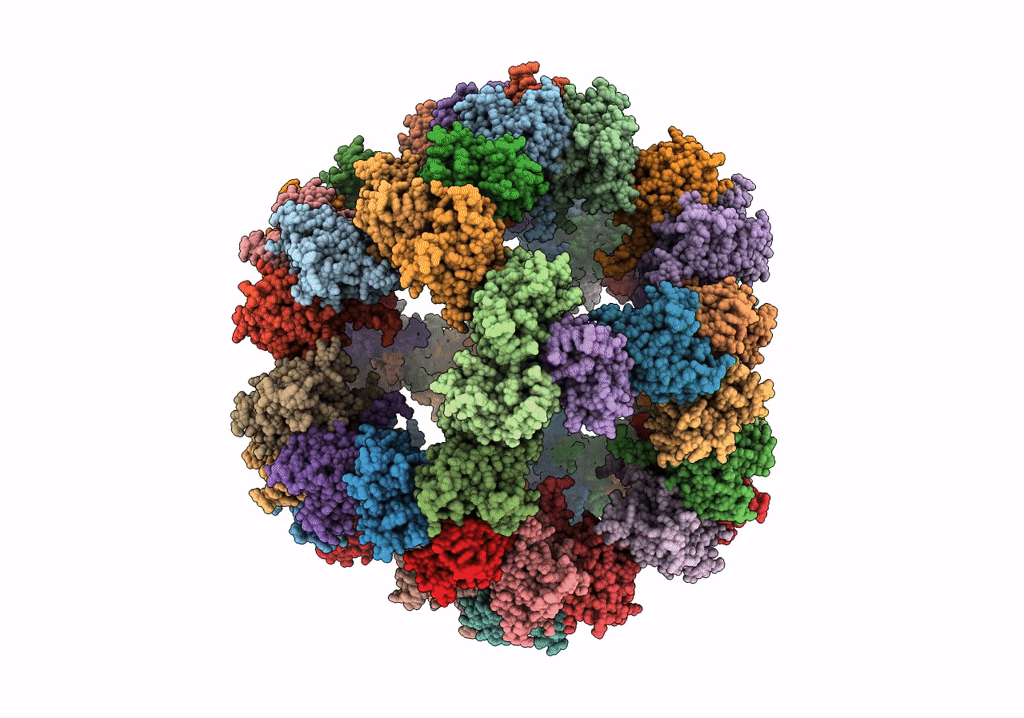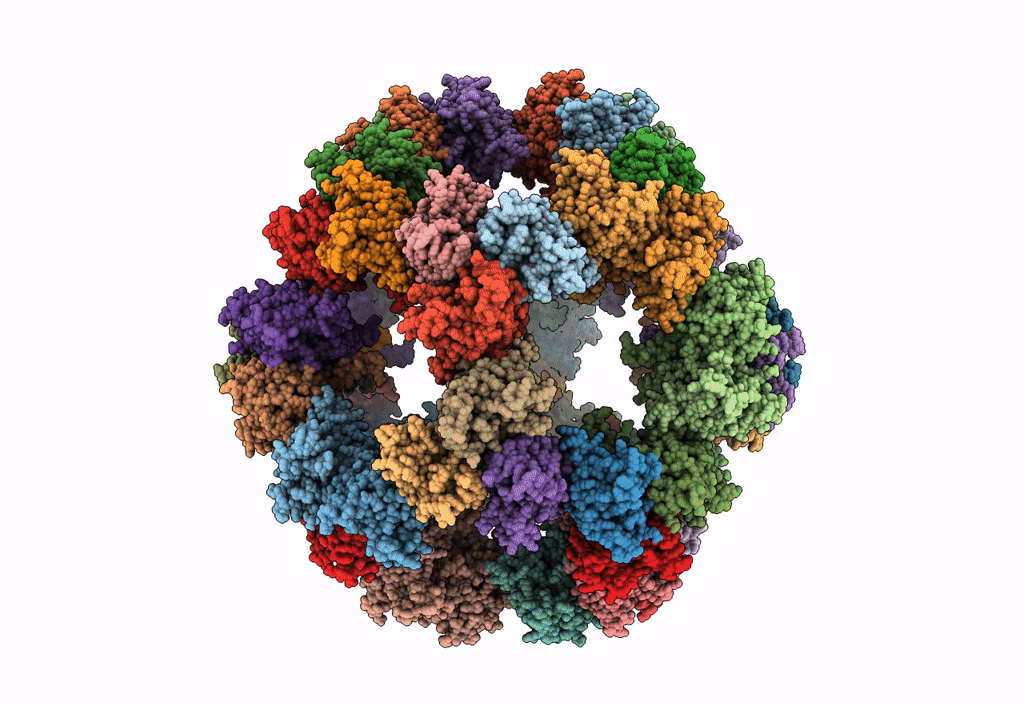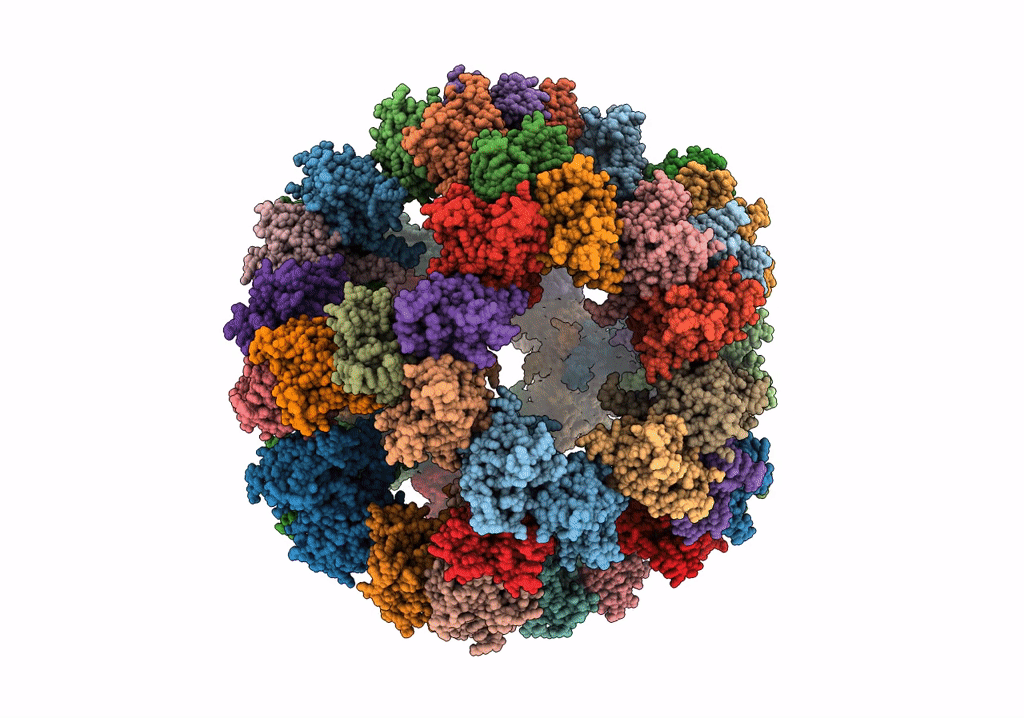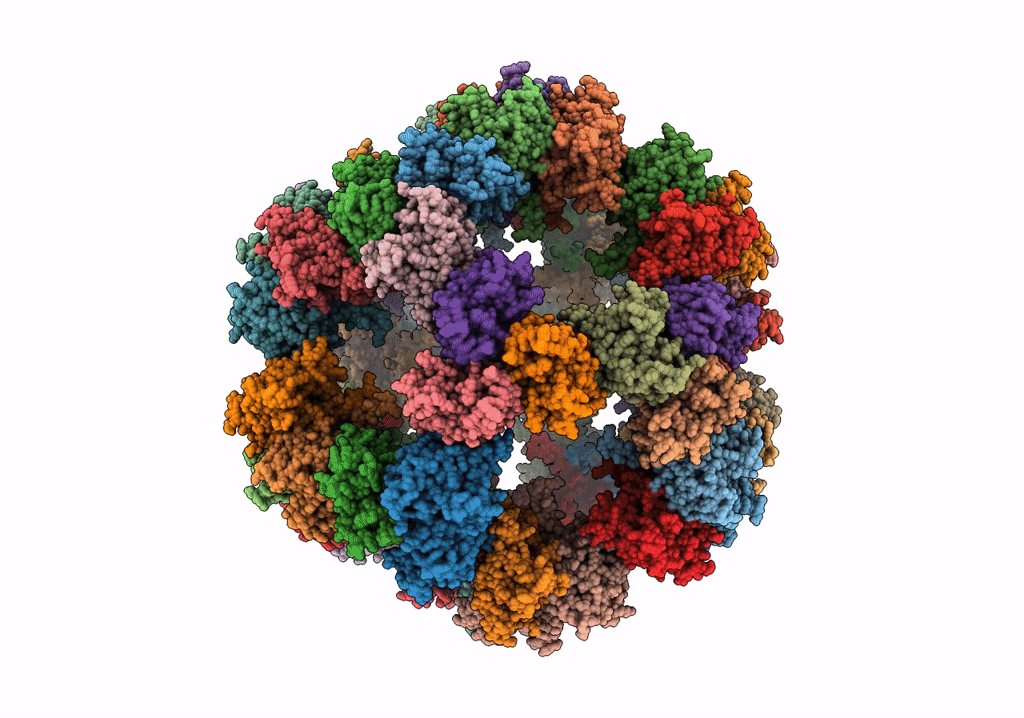
PDB ID:
7Q5R
Keywords:
Title:
Protein community member pyruvate dehydrogenase complex E2 core from C. thermophilum
Biological Source:
Source Organism(s):
Method Details:
Experimental Method:
Resolution:
3.84 Å
Aggregation State:
PARTICLE
Reconstruction Method:
SINGLE PARTICLE