Deposition Date
2021-02-20
Release Date
2022-08-17
Last Version Date
2024-10-30
Entry Detail
PDB ID:
7LU0
Keywords:
Title:
X-ray radiation damage series on Proteinase K at 100K, crystal structure, dataset 4
Biological Source:
Source Organism(s):
Parengyodontium album (Taxon ID: 37998)
Method Details:
Experimental Method:
Resolution:
1.01 Å
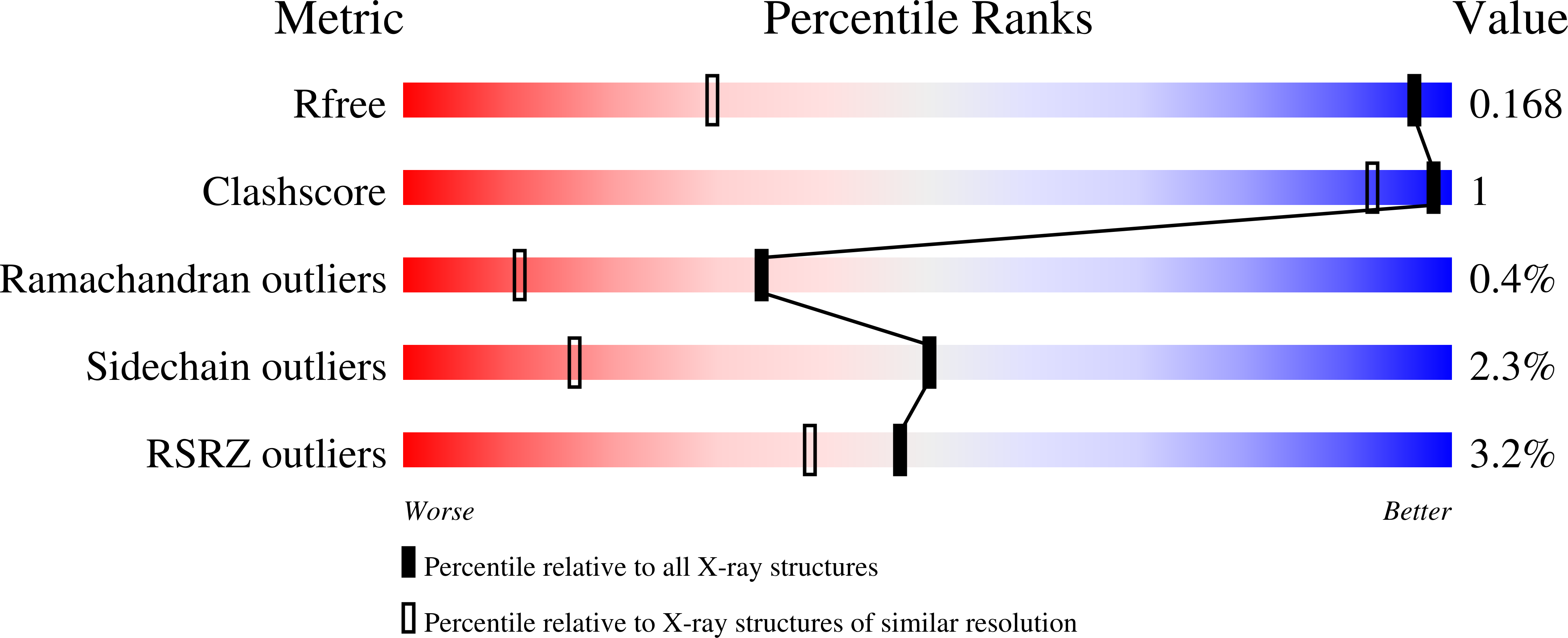
R-Value Free:
0.16
R-Value Work:
0.14
R-Value Observed:
0.14
Space Group:
P 43 21 2