Deposition Date
2020-03-26
Release Date
2021-03-31
Last Version Date
2023-12-20
Entry Detail
PDB ID:
7BR4
Keywords:
Title:
Structure of deletion mutant of alpha-glucuronidase (TM0752) from Thermotoga maritima
Biological Source:
Source Organism(s):
Expression System(s):
Method Details:
Experimental Method:
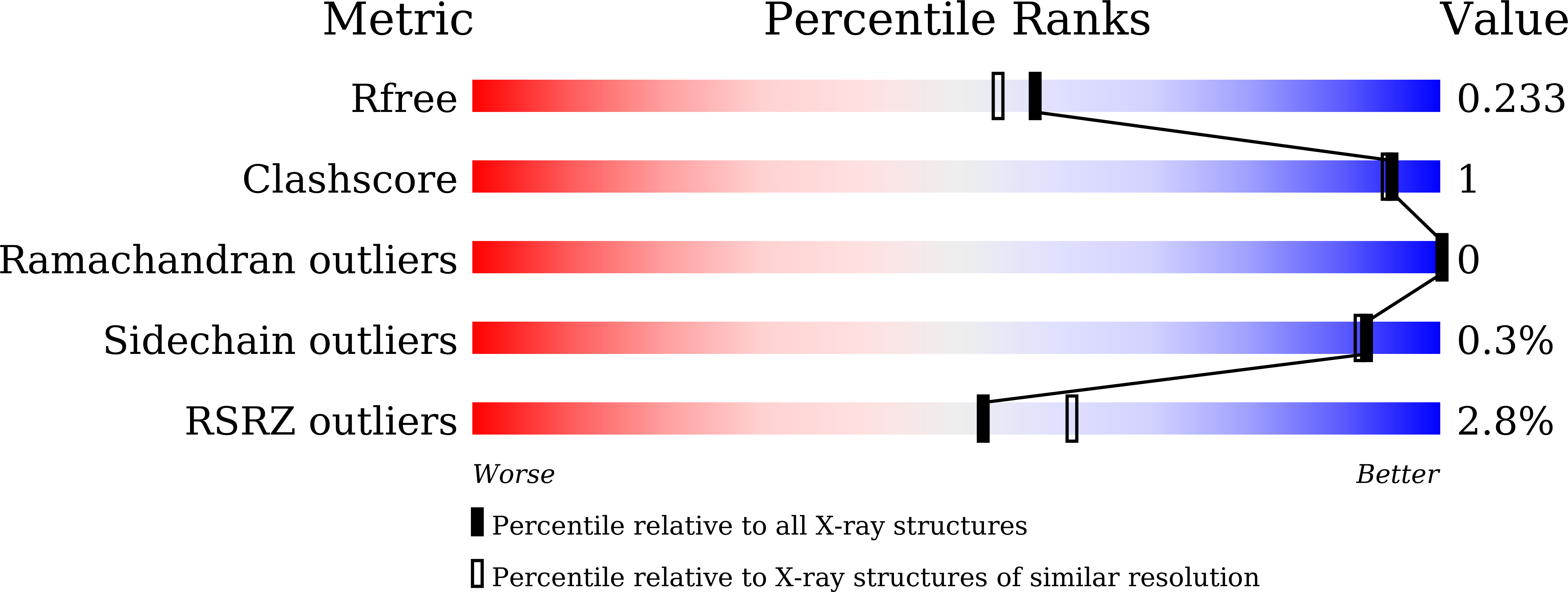
Resolution:
1.95 Å
R-Value Free:
0.23
R-Value Work:
0.19
R-Value Observed:
0.19
Space Group:
C 1 2 1