
Deposition Date
2020-01-31
Release Date
2021-02-10
Last Version Date
2024-01-24
Entry Detail
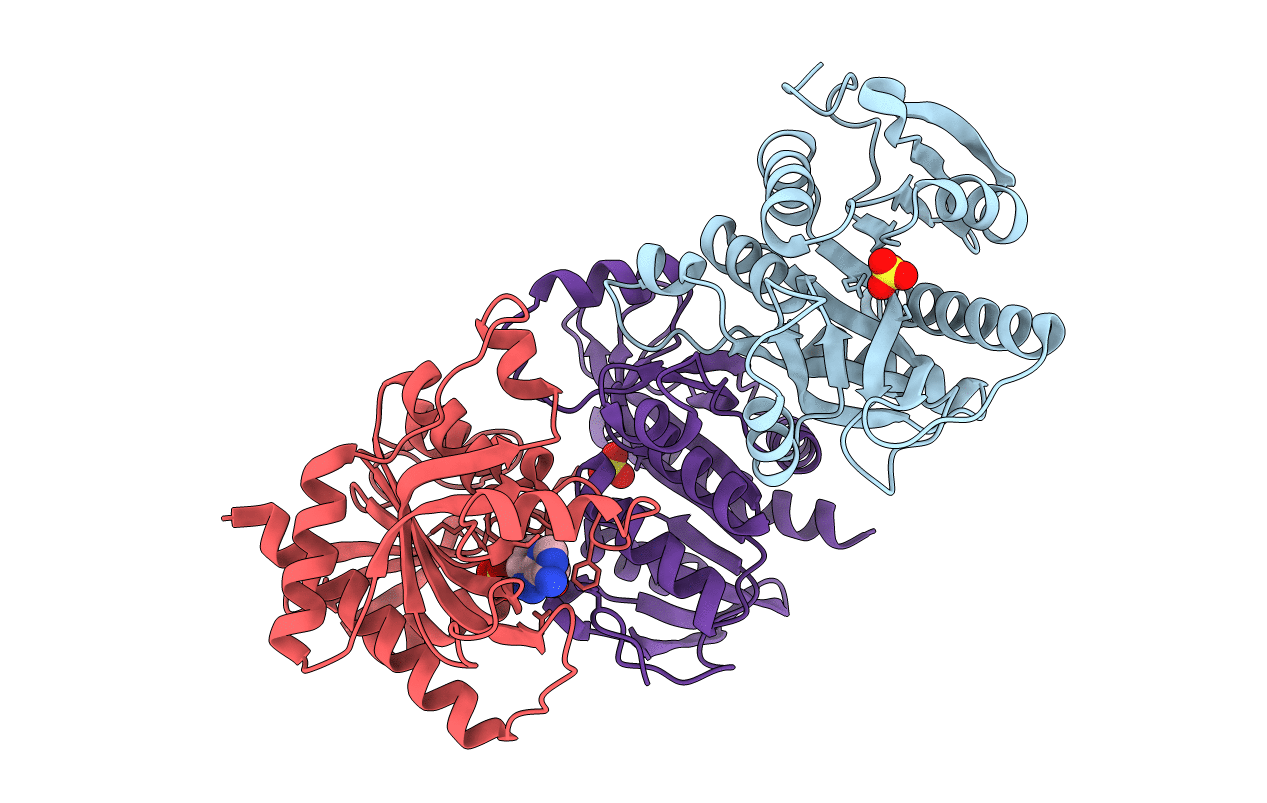
PDB ID:
6XZ2
Keywords:
Title:
Crystal structure of E. Coli purine nucleoside phosphorylase mutant Y160W with SO4 and Formycin A
Biological Source:
Source Organism(s):
Escherichia coli (strain K12) (Taxon ID: 83333)
Expression System(s):
Method Details:
Experimental Method:
Resolution:
1.65 Å
R-Value Free:
0.19
R-Value Work:
0.17
R-Value Observed:
0.18
Space Group:
P 61 2 2
