Deposition Date
2019-02-10
Release Date
2019-11-13
Last Version Date
2024-10-30
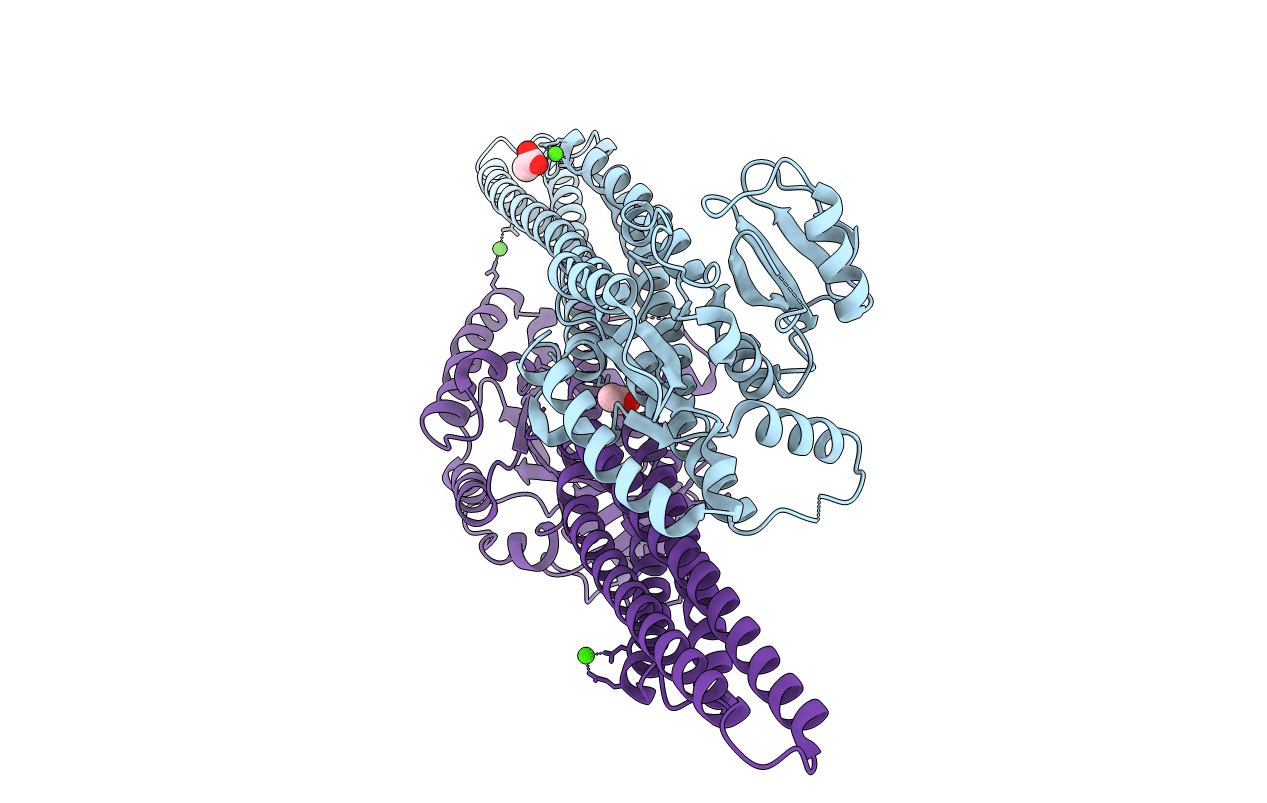
Entry Detail
Biological Source:
Source Organism(s):
Homo sapiens (Taxon ID: 9606)
Macaca mulatta (Taxon ID: 9544)
Macaca mulatta (Taxon ID: 9544)
Expression System(s):
Method Details:
Experimental Method:
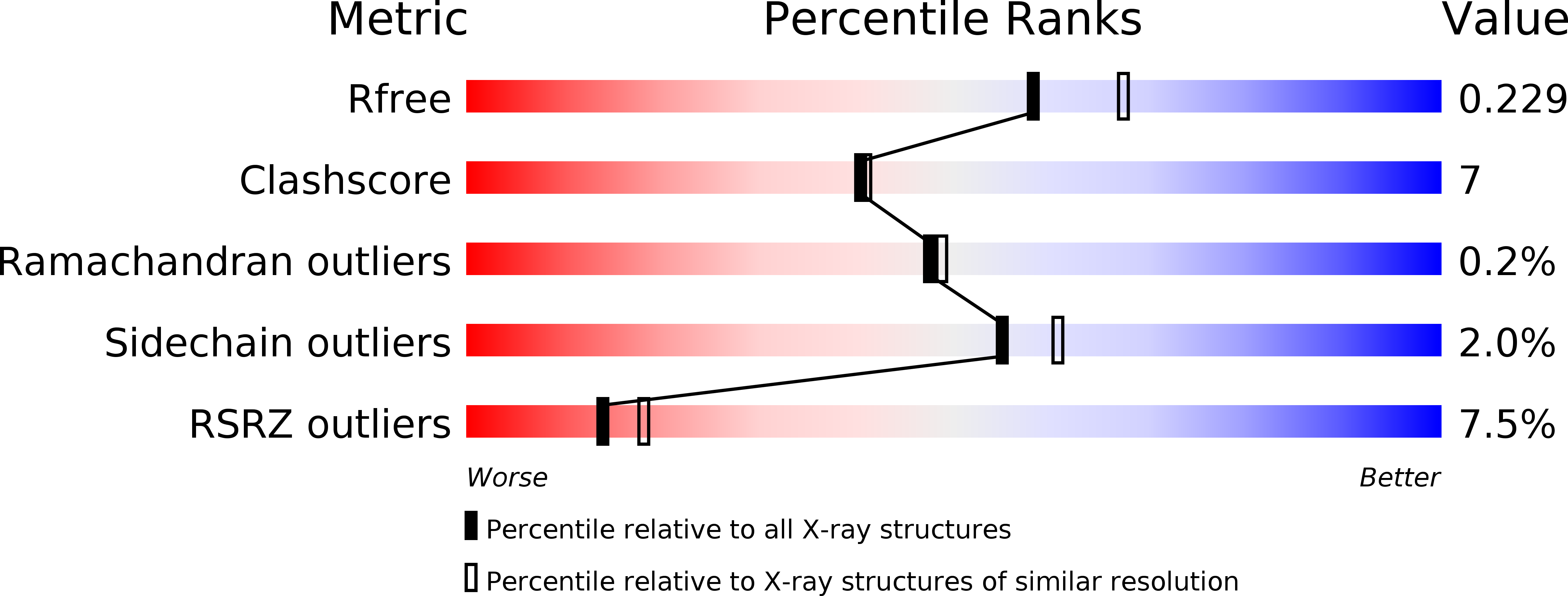
Resolution:
2.09 Å
R-Value Free:
0.22
R-Value Work:
0.17
R-Value Observed:
0.17
Space Group:
P 1 21 1