Deposition Date
2018-11-15
Release Date
2019-11-27
Last Version Date
2024-01-24
Entry Detail
PDB ID:
6I6Y
Keywords:
Title:
Circular permutant of ribosomal protein S6, swap helix 2
Biological Source:
Source Organism:
Thermus thermophilus HB8 (Taxon ID: 300852)
Host Organism:
Method Details:
Experimental Method:
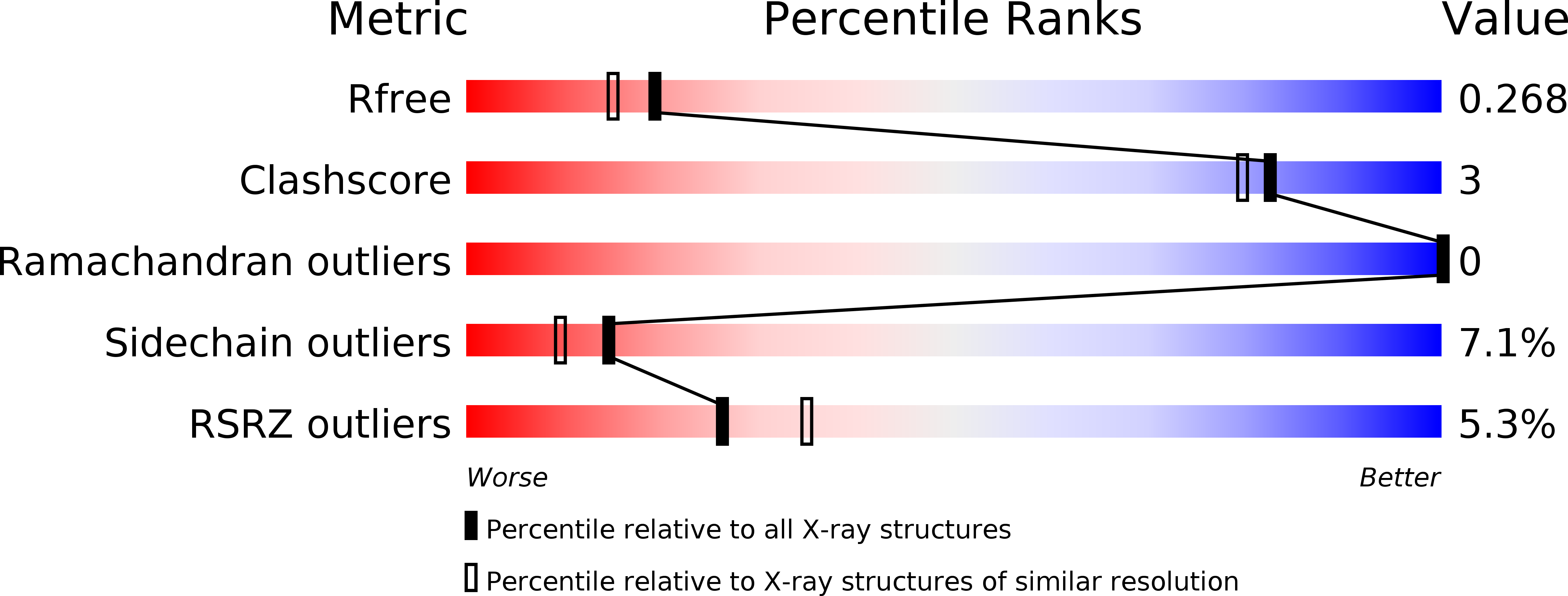
Resolution:
2.15 Å
R-Value Free:
0.26
R-Value Work:
0.22
R-Value Observed:
0.23
Space Group:
P 41 21 2