Deposition Date
2008-09-25
Release Date
2008-10-07
Last Version Date
2024-11-20
Entry Detail
PDB ID:
3EMY
Keywords:
Title:
Crystal structure of Trichoderma reesei aspartic proteinase complexed with pepstatin A
Biological Source:
Source Organism(s):
Streptomyces argenteolus subsp. toyonakensis (Taxon ID: 285516)
Hypocrea jecorina (Taxon ID: 51453)
Hypocrea jecorina (Taxon ID: 51453)
Method Details:
Experimental Method:
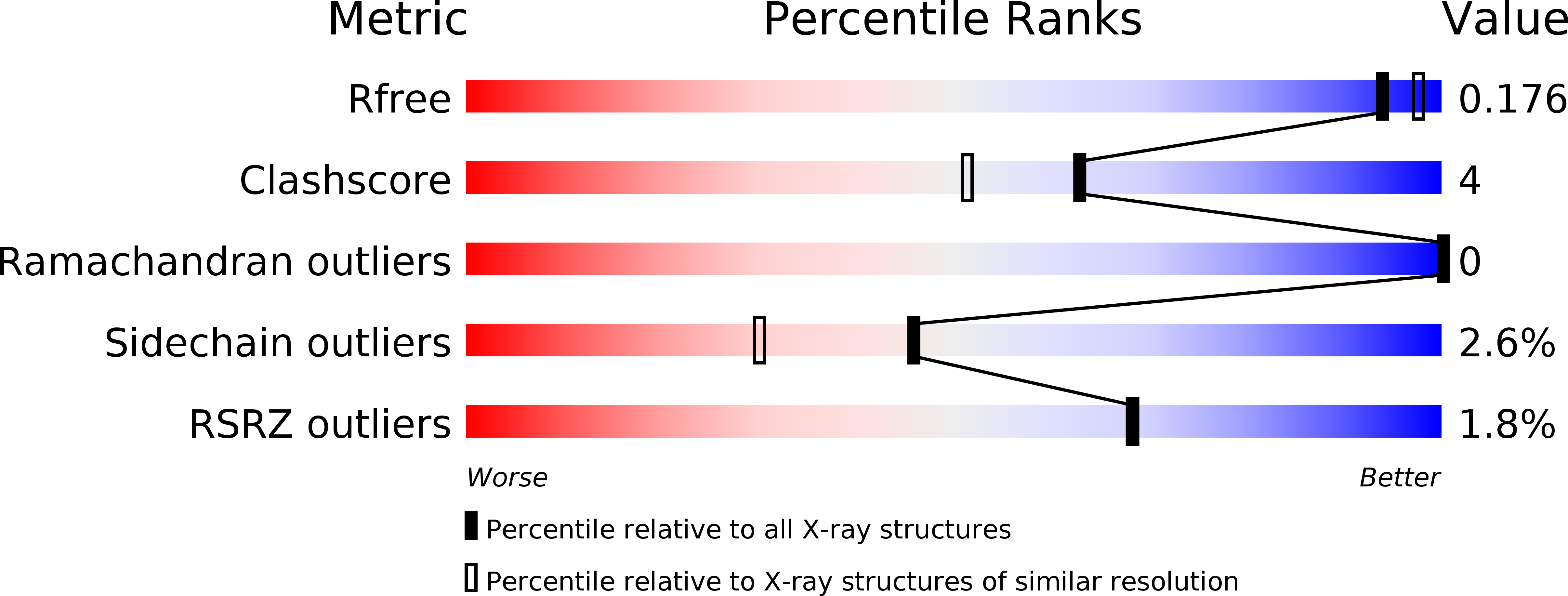
Resolution:
1.85 Å
R-Value Free:
0.18
R-Value Work:
0.14
R-Value Observed:
0.14
Space Group:
P 43 21 2