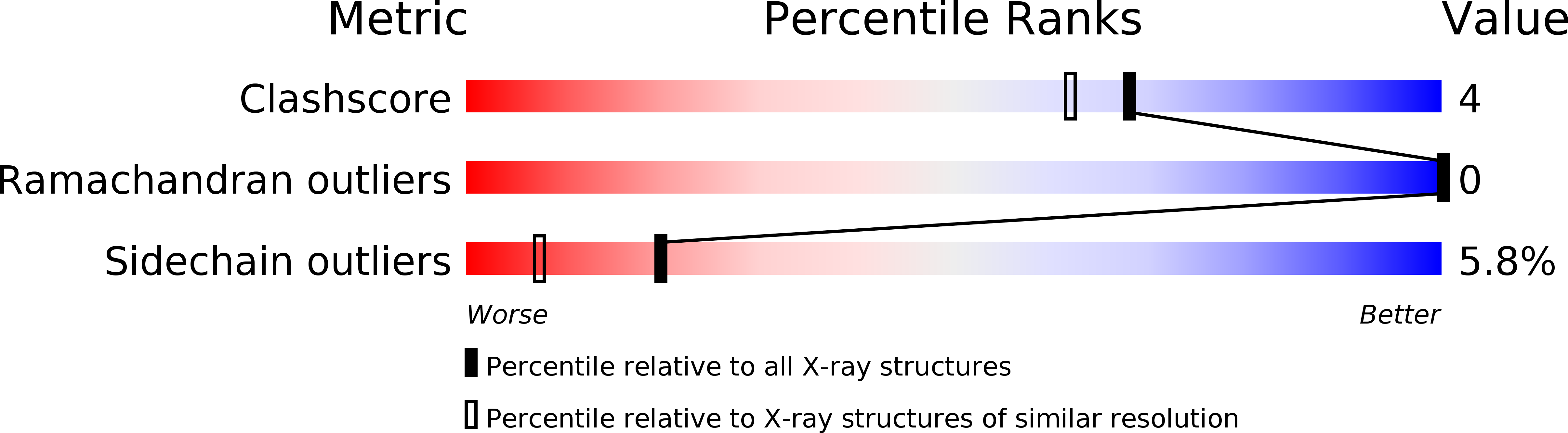Deposition Date
1994-07-11
Release Date
1995-02-07
Last Version Date
2024-10-23
Entry Detail
PDB ID:
2EXO
Keywords:
Title:
CRYSTAL STRUCTURE OF THE CATALYTIC DOMAIN OF THE BETA-1,4-GLYCANASE CEX FROM CELLULOMONAS FIMI
Biological Source:
Source Organism(s):
Cellulomonas fimi (Taxon ID: 1708)
Method Details:
Experimental Method:
Resolution:
1.80 Å
R-Value Free:
0.26
R-Value Work:
0.21
R-Value Observed:
0.21
Space Group:
P 41 21 2