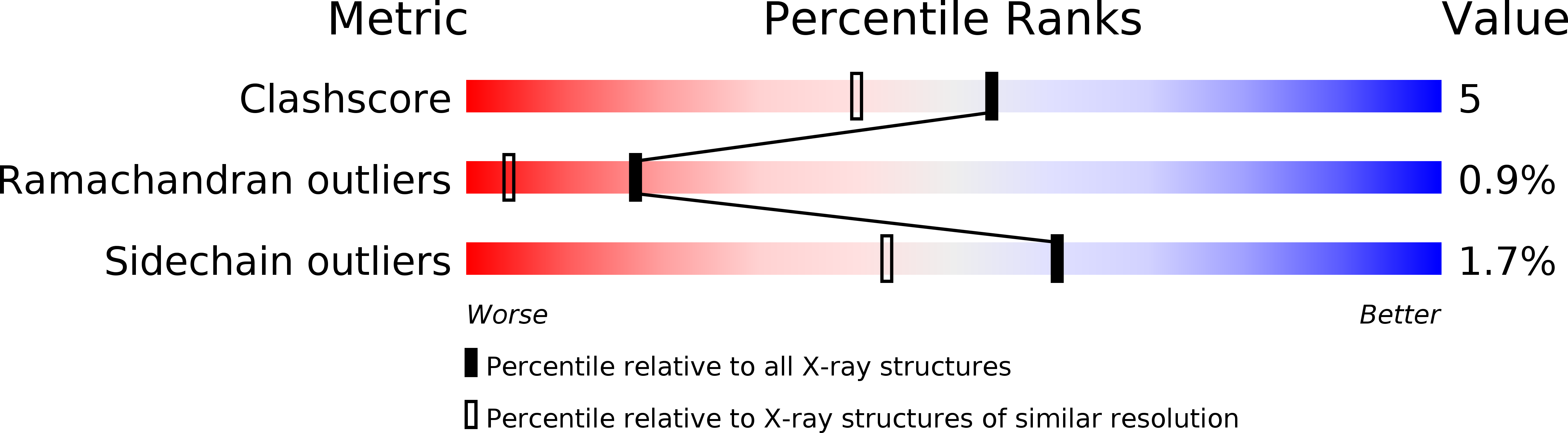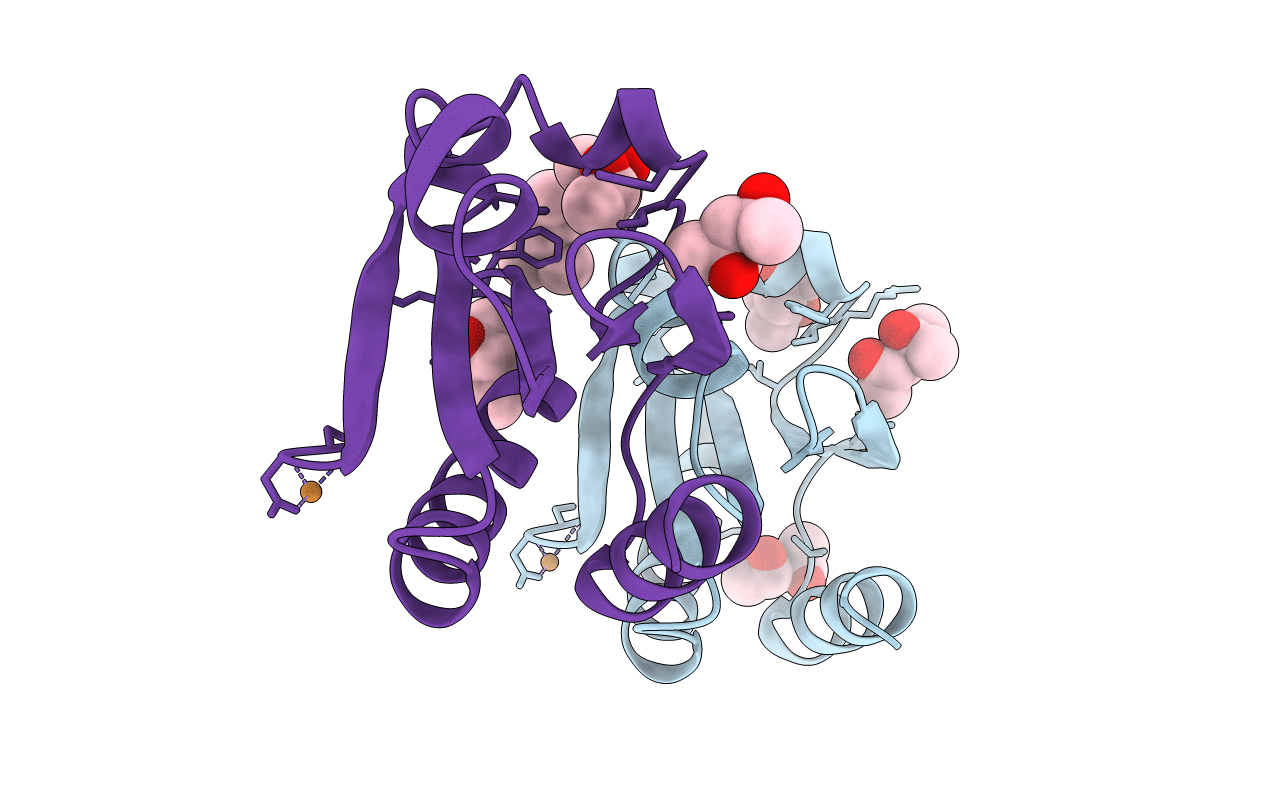
Deposition Date
1990-03-19
Release Date
1991-10-15
Last Version Date
2024-11-20
Entry Detail
PDB ID:
2TRX
Keywords:
Title:
CRYSTAL STRUCTURE OF THIOREDOXIN FROM ESCHERICHIA COLI AT 1.68 ANGSTROMS RESOLUTION
Biological Source:
Source Organism(s):
Escherichia coli (Taxon ID: 562)
Method Details:
Experimental Method:
Resolution:
1.68 Å
R-Value Observed:
0.16
Space Group:
C 1 2 1