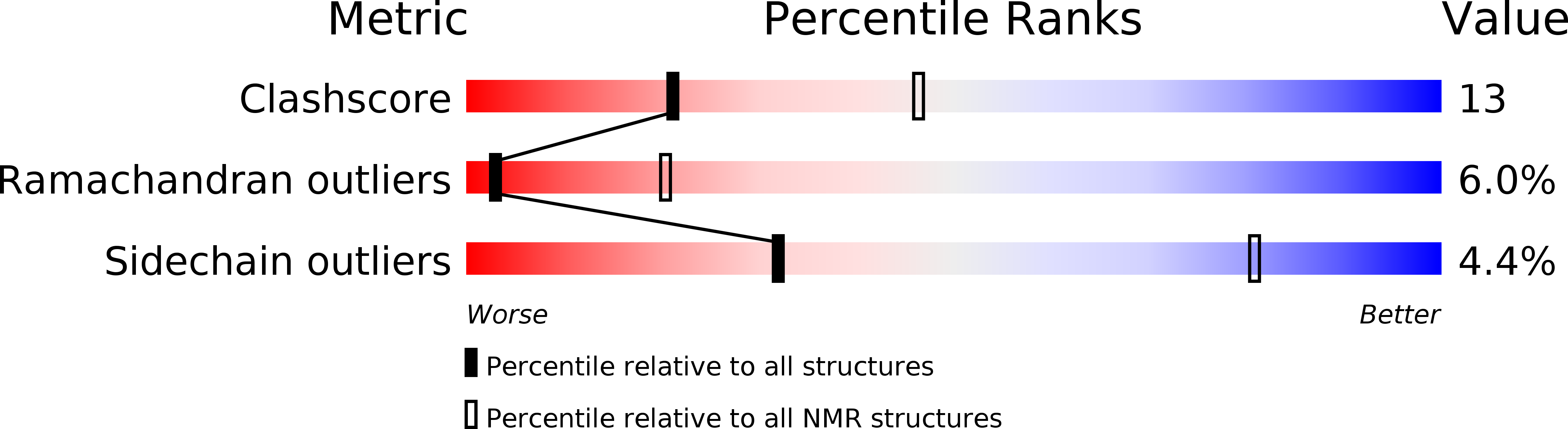
Deposition Date
2011-05-10
Release Date
2011-06-29
Last Version Date
2024-05-15
Entry Detail
PDB ID:
2LCV
Keywords:
Title:
Structure of the Cytidine Repressor DNA-Binding Domain; an alternate calculation
Biological Source:
Source Organism(s):
Escherichia coli K-12 (Taxon ID: 83333)
Expression System(s):
Method Details:
Experimental Method:
Conformers Calculated:
100
Conformers Submitted:
11
Selection Criteria:
structures with the lowest energy