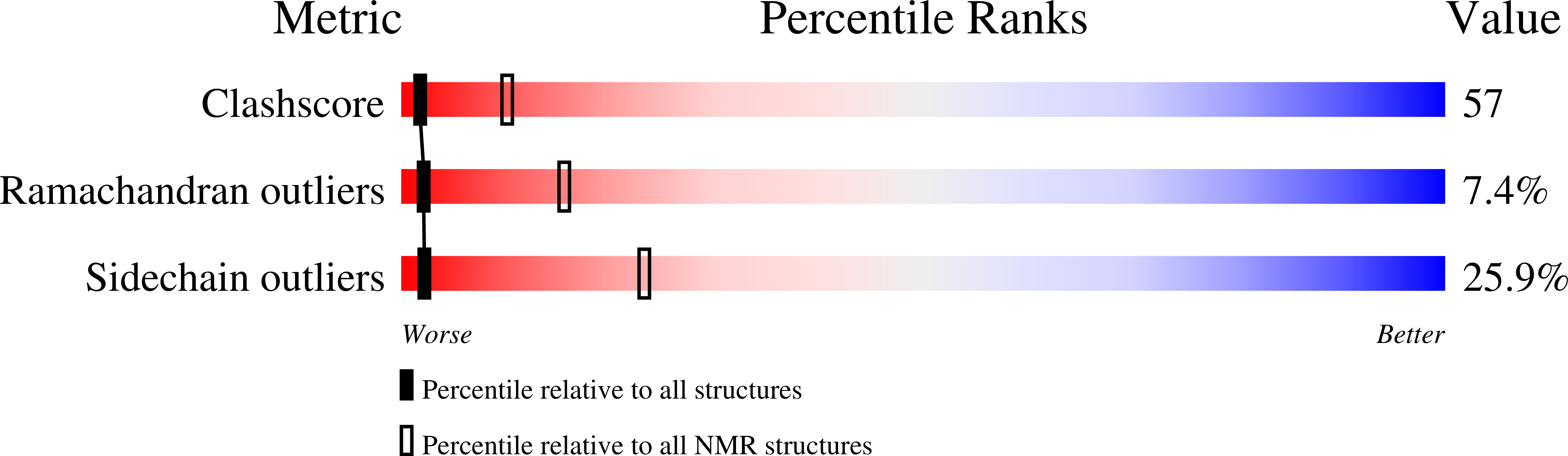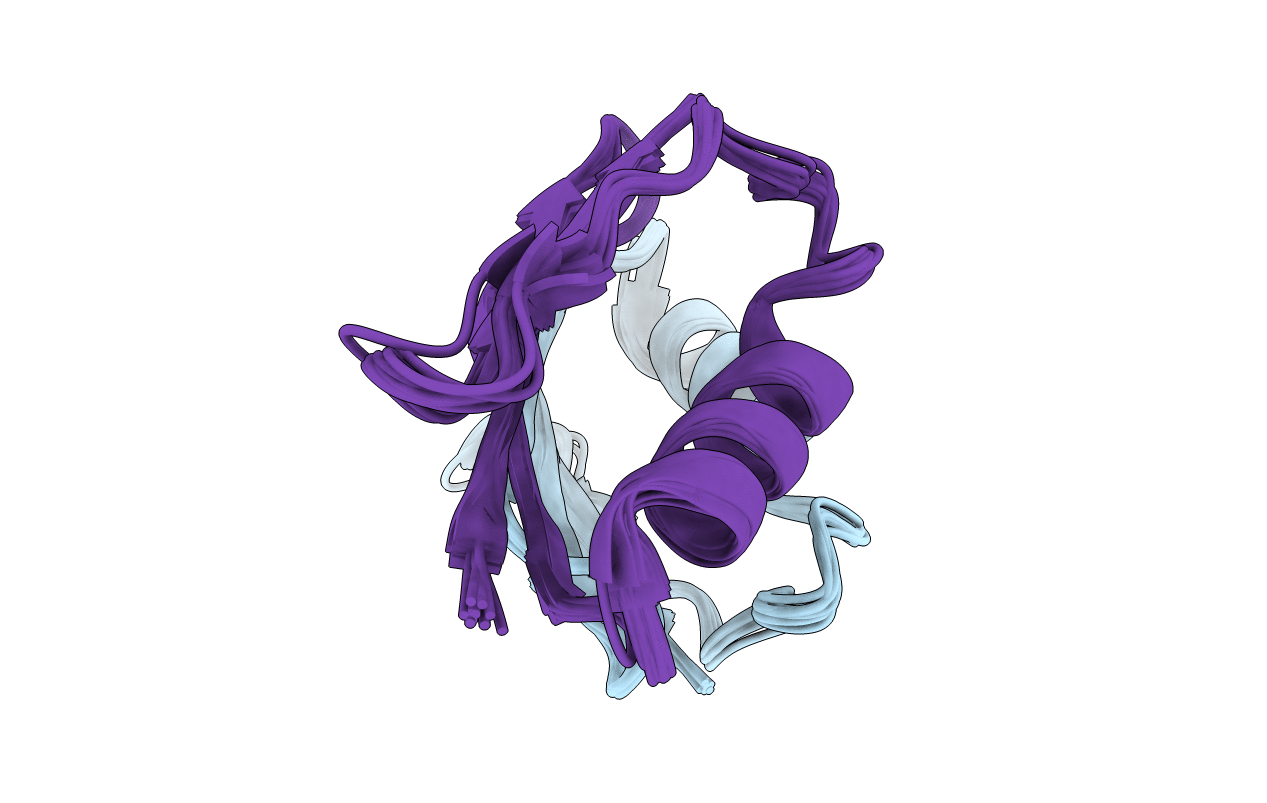
Deposition Date
2009-07-06
Release Date
2009-10-06
Last Version Date
2024-05-22
Entry Detail
Biological Source:
Source Organism(s):
Staphylococcus aureus (Taxon ID: 1280)
Expression System(s):
Method Details:
Experimental Method:
Conformers Calculated:
100
Conformers Submitted:
10
Selection Criteria:
structures with the lowest energy