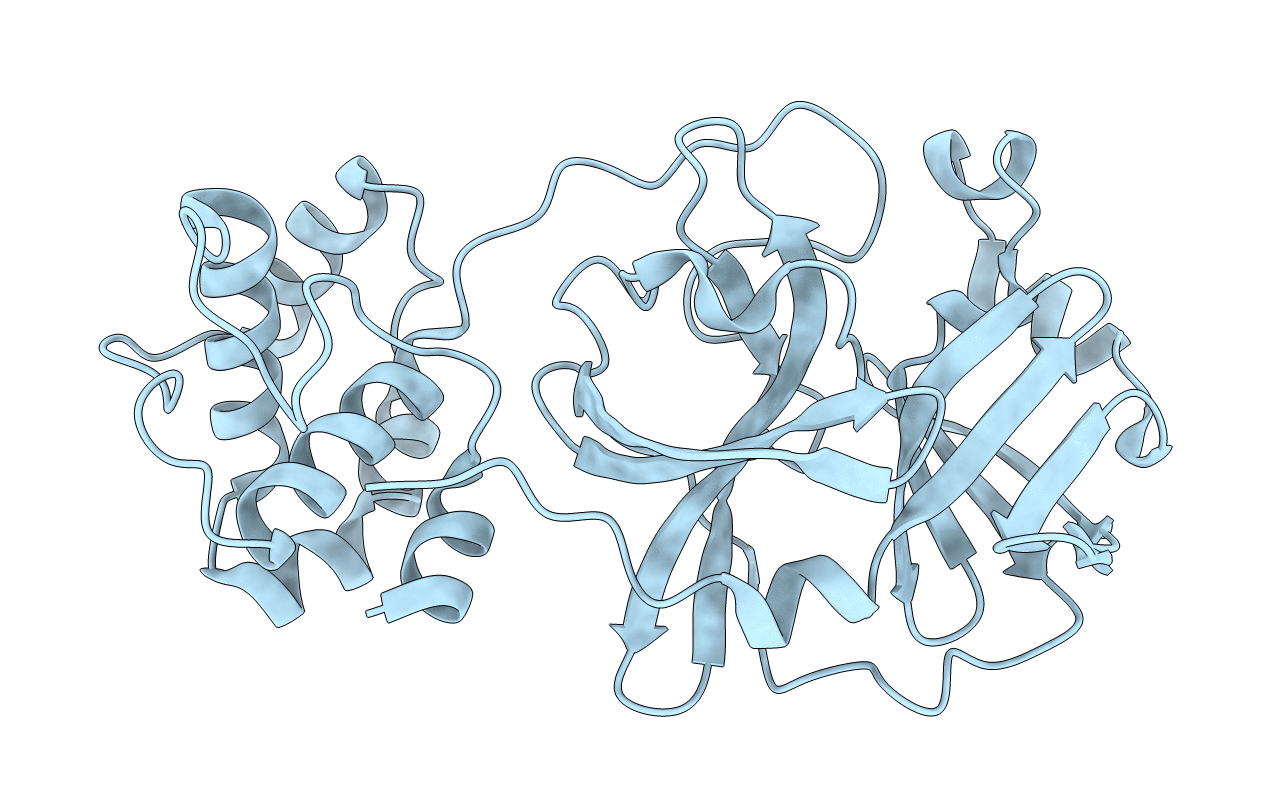
Deposition Date
2006-04-27
Release Date
2006-12-26
Last Version Date
2024-02-14
Entry Detail
PDB ID:
2GT8
Keywords:
Title:
Crystal structure of SARS coronavirus main peptidase (with an additional Ala at the N-terminus of each protomer) in the space group P43212
Biological Source:
Source Organism(s):
SARS coronavirus (Taxon ID: 227859)
Expression System(s):
Method Details:
Experimental Method:
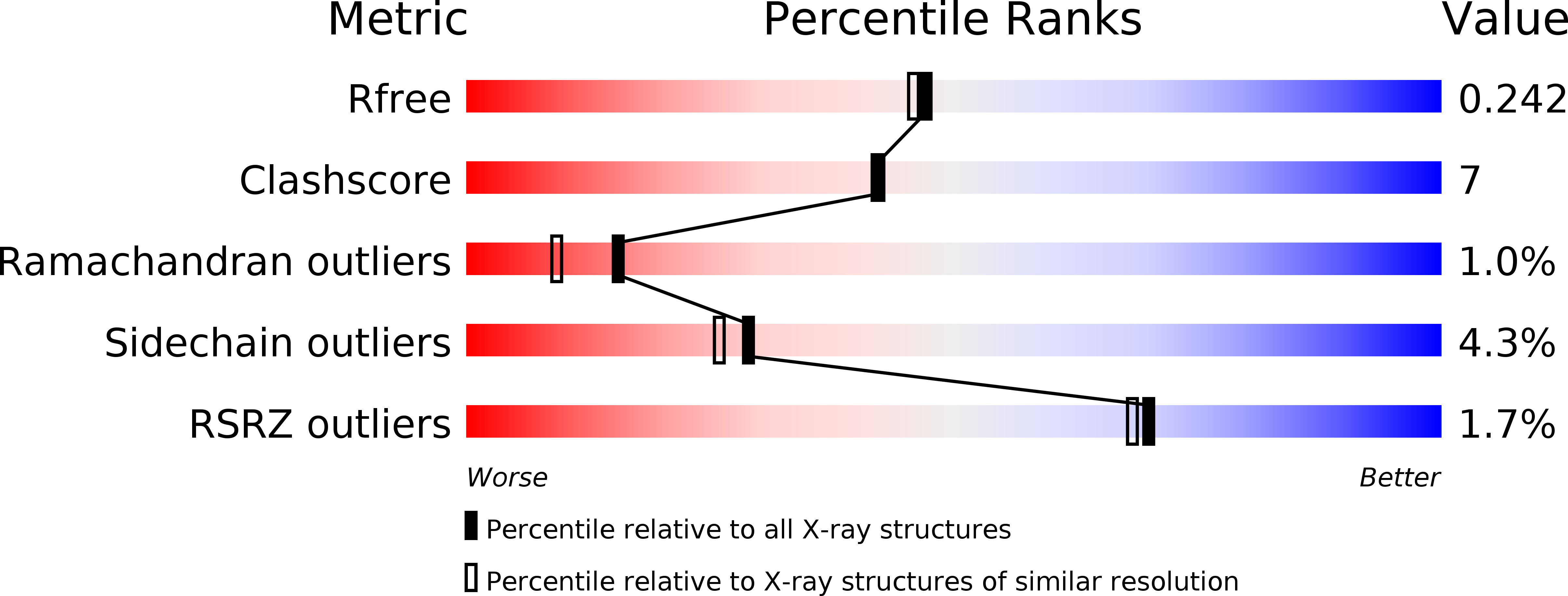
Resolution:
2.00 Å
R-Value Free:
0.24
R-Value Work:
0.17
R-Value Observed:
0.17
Space Group:
P 43 21 2