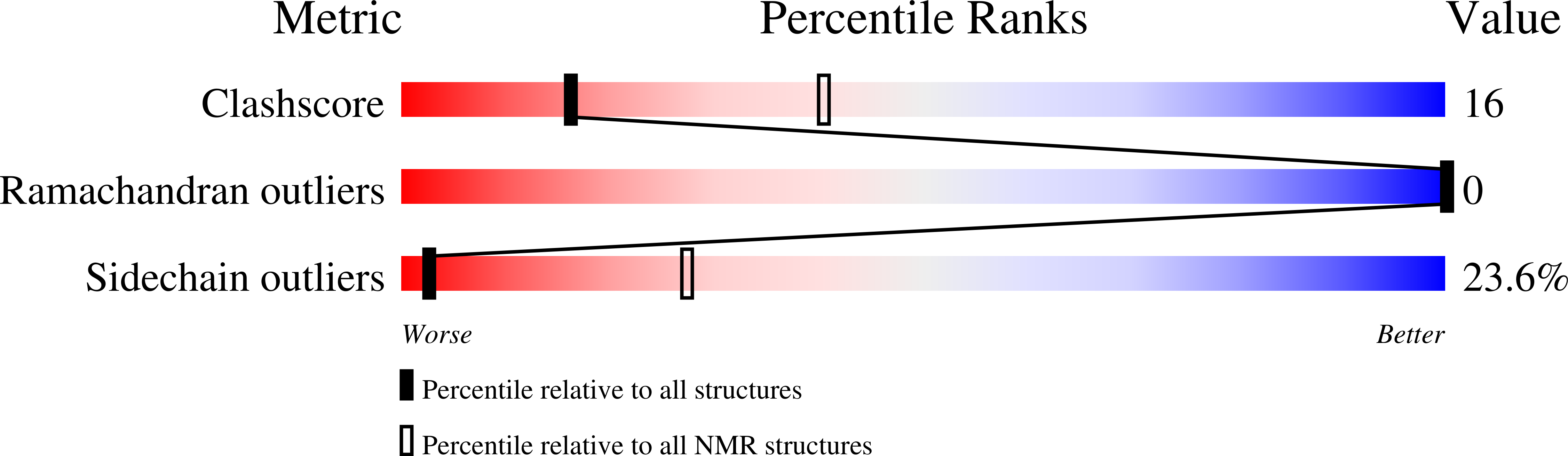
Deposition Date
2006-03-30
Release Date
2006-10-17
Last Version Date
2024-05-29
Entry Detail
PDB ID:
2GJH
Keywords:
Title:
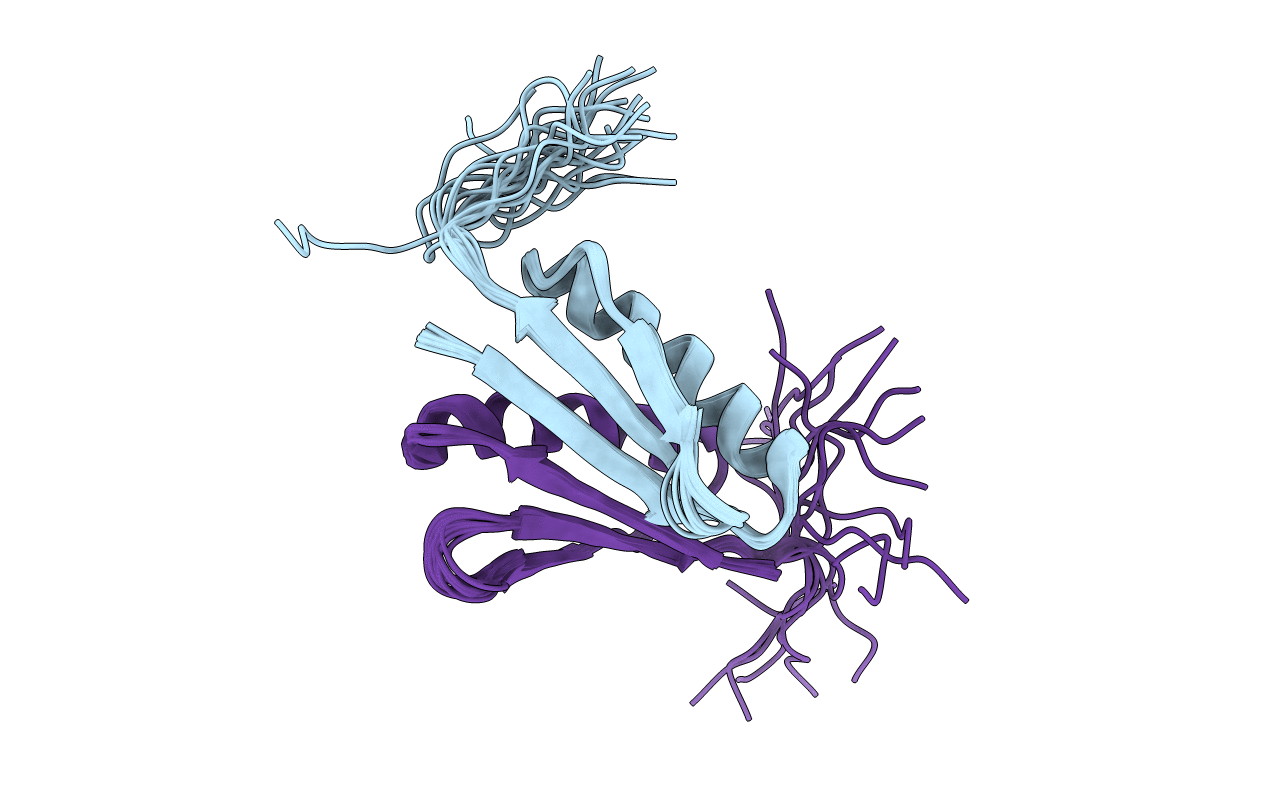
NMR Structure of CFr (C-terminal fragment of computationally designed novel-topology protein Top7)
Biological Source:
Expression System(s):
Method Details:
Experimental Method:
Conformers Calculated:
100
Conformers Submitted:
20
Selection Criteria:
structures with the least restraint violations