Deposition Date
2004-11-30
Release Date
2005-05-17
Last Version Date
2023-08-23
Entry Detail
PDB ID:
1Y4D
Keywords:
Title:
Crystal structure of the complex of subtilisin BPN' with chymotrypsin inhibitor 2 M59R/E60S mutant
Biological Source:
Source Organism(s):
Bacillus amyloliquefaciens (Taxon ID: 1390)
Hordeum vulgare (Taxon ID: 4513)
Hordeum vulgare (Taxon ID: 4513)
Expression System(s):
Method Details:
Experimental Method:
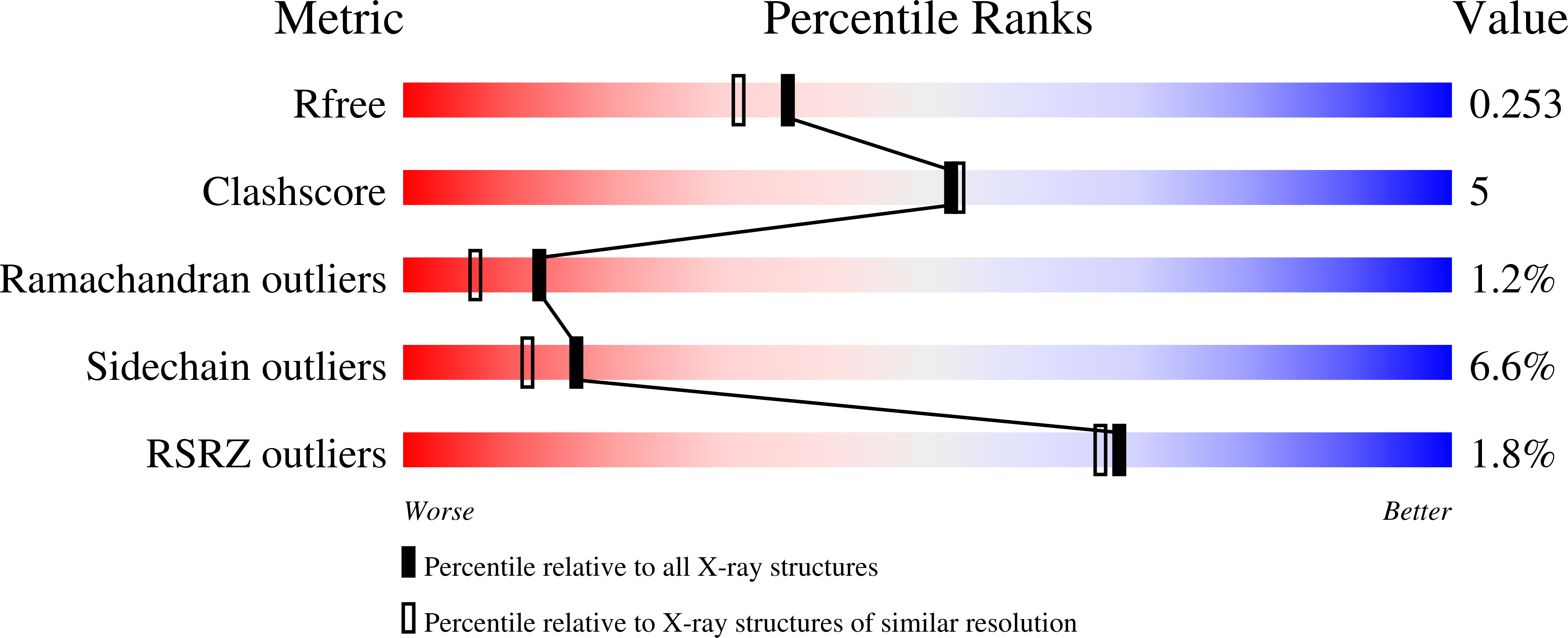
Resolution:
2.00 Å
R-Value Free:
0.25
R-Value Work:
0.19
R-Value Observed:
0.19
Space Group:
P 61