Deposition Date
2003-10-15
Release Date
2004-07-06
Last Version Date
2023-12-27
Entry Detail
PDB ID:
1V2E
Keywords:
Title:
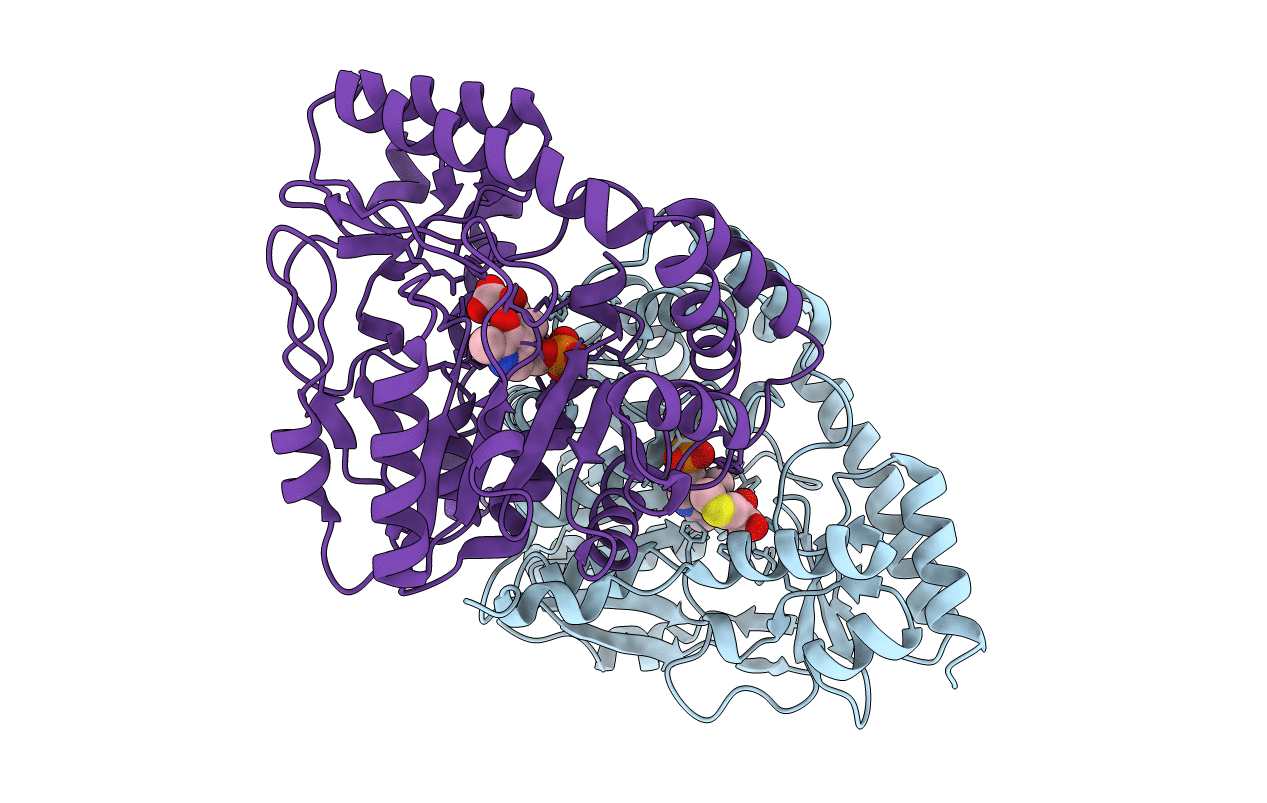
Crystal Structure of T.th HB8 Glutamine Aminotransferase complex with a-keto-g-methylthiobutyrate
Biological Source:
Source Organism(s):
Thermus thermophilus (Taxon ID: 274)
Expression System(s):
Method Details:
Experimental Method:
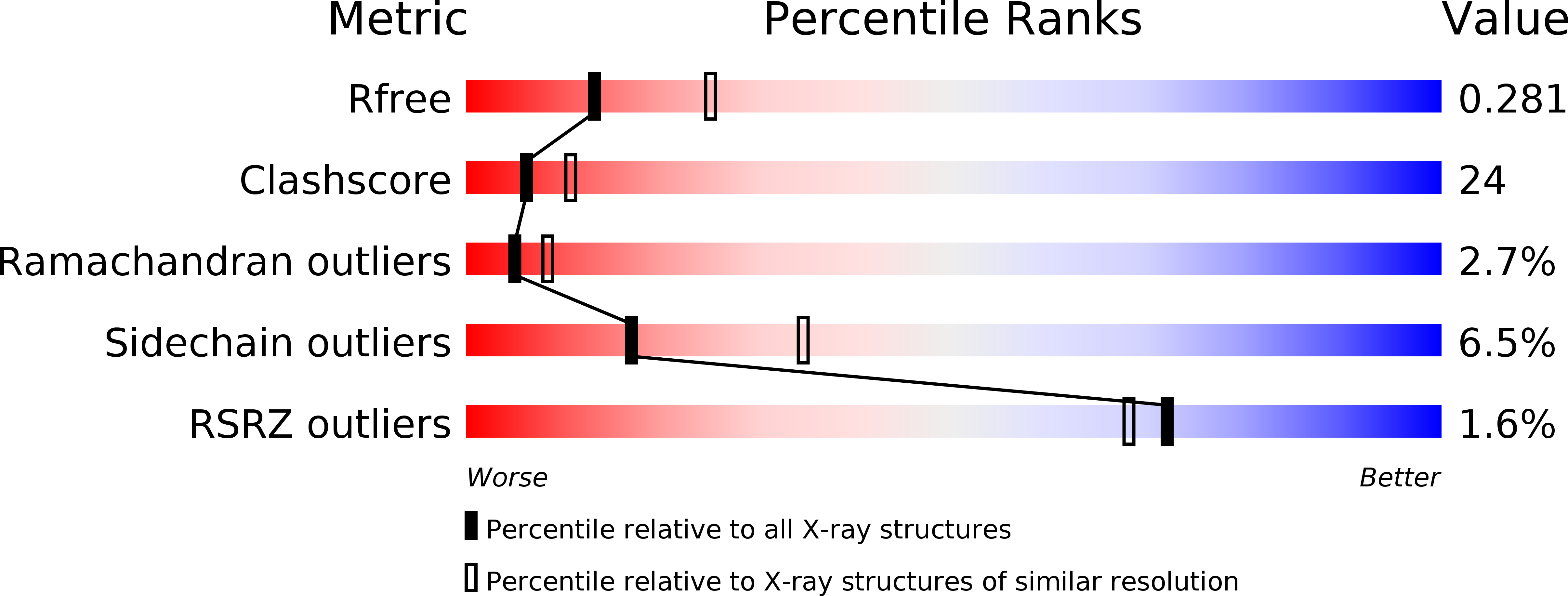
Resolution:
2.60 Å
R-Value Free:
0.28
R-Value Work:
0.23
Space Group:
P 31 2 1