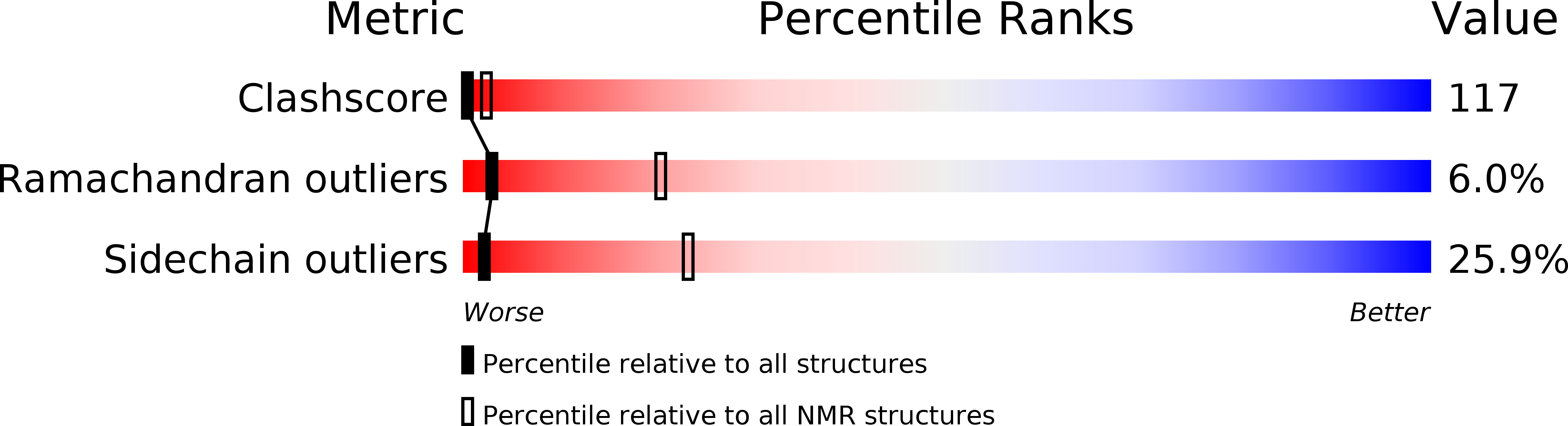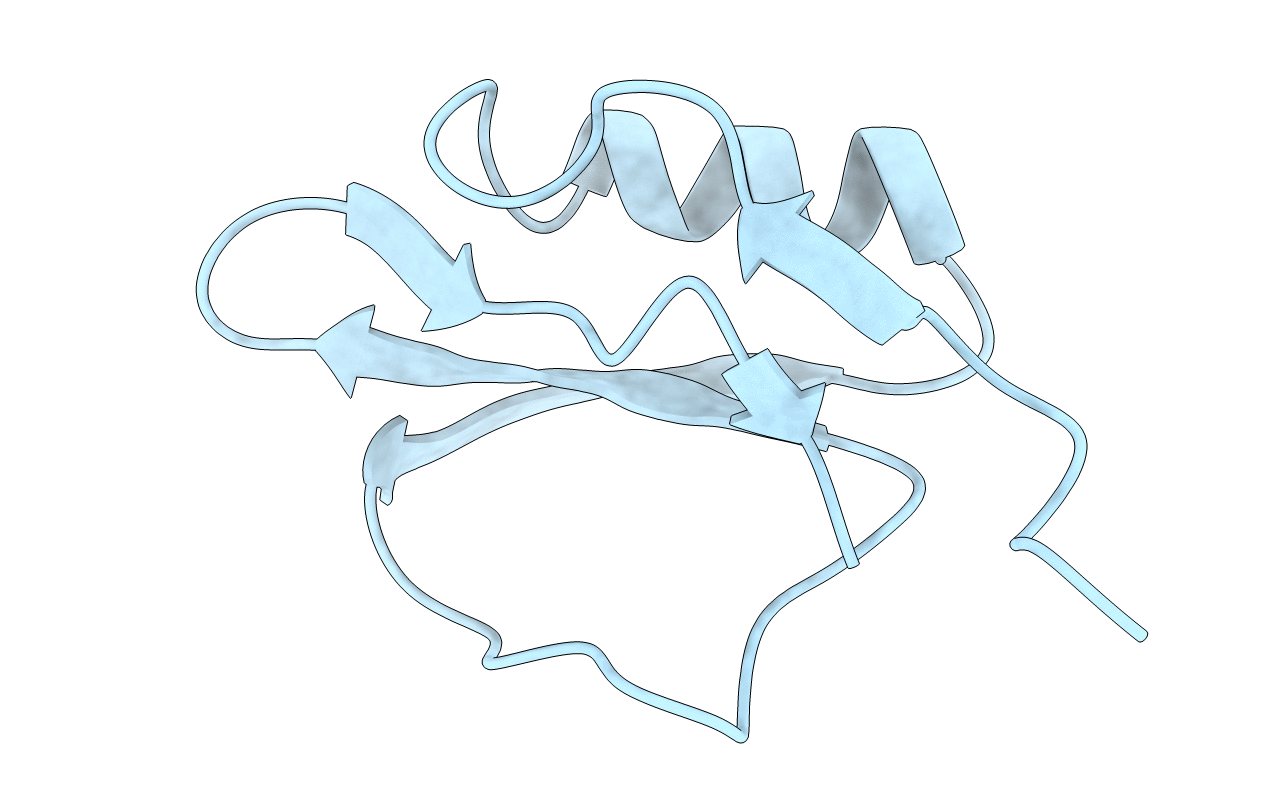
Deposition Date
1994-10-28
Release Date
1995-01-26
Last Version Date
2024-11-20
Entry Detail
PDB ID:
1TIN
Keywords:
Title:
THREE-DIMENSIONAL STRUCTURE IN SOLUTION OF CUCURBITA MAXIMA TRYPSIN INHIBITOR-V DETERMINED BY NMR SPECTROSCOPY
Biological Source:
Source Organism(s):
Cucurbita maxima (Taxon ID: 3661)
Method Details:
Experimental Method:
Conformers Submitted:
1