Deposition Date
2003-07-23
Release Date
2003-08-19
Last Version Date
2024-11-20
Entry Detail
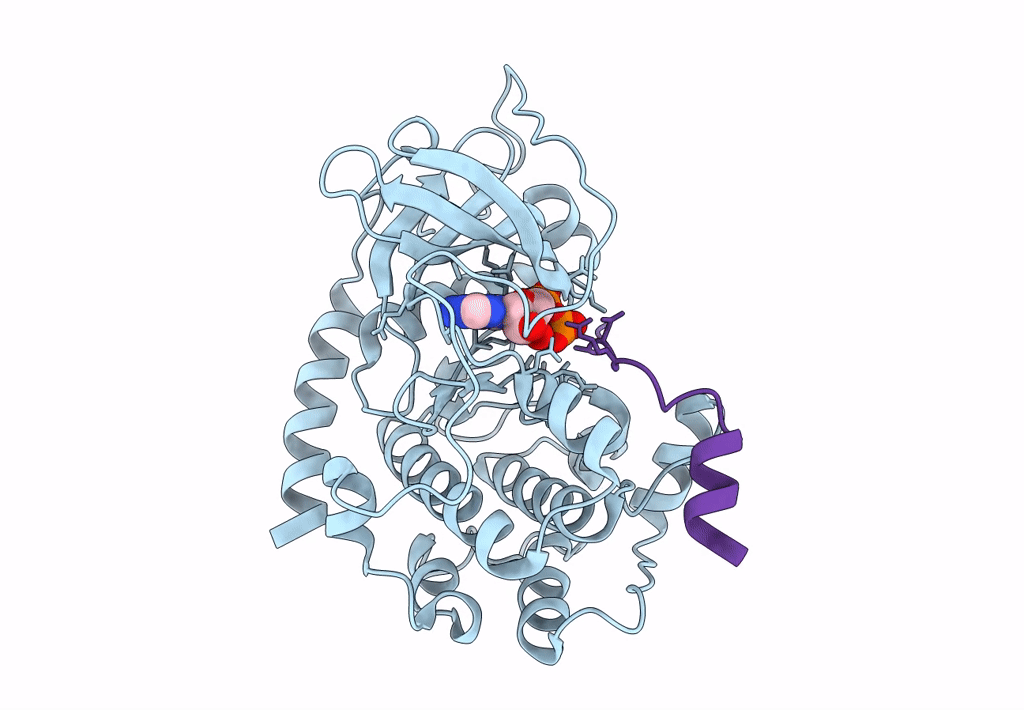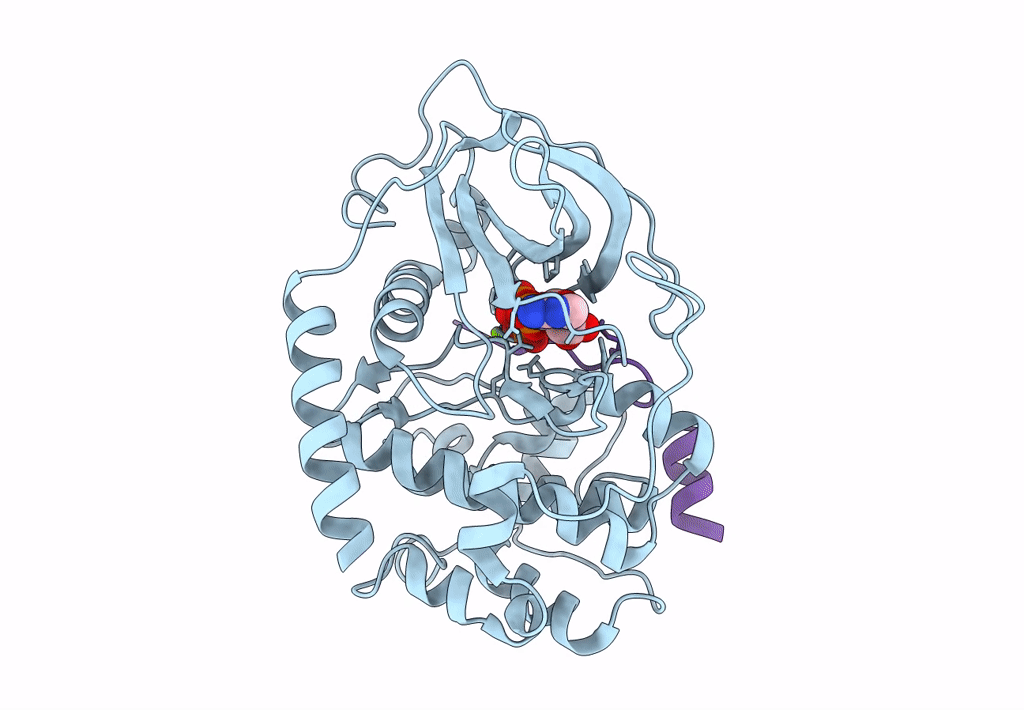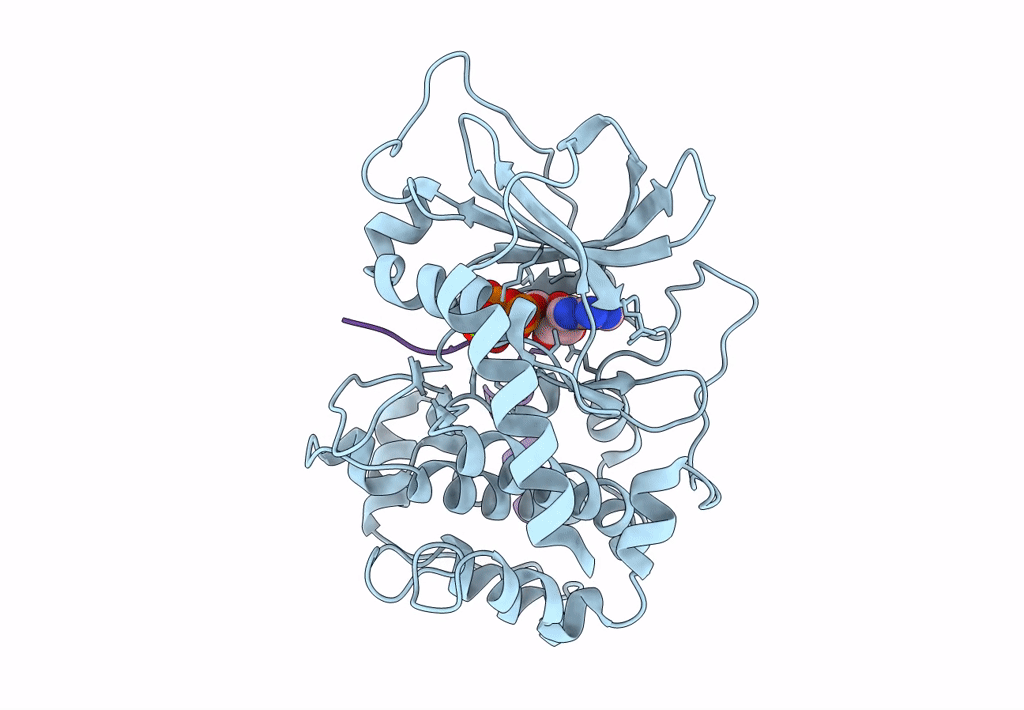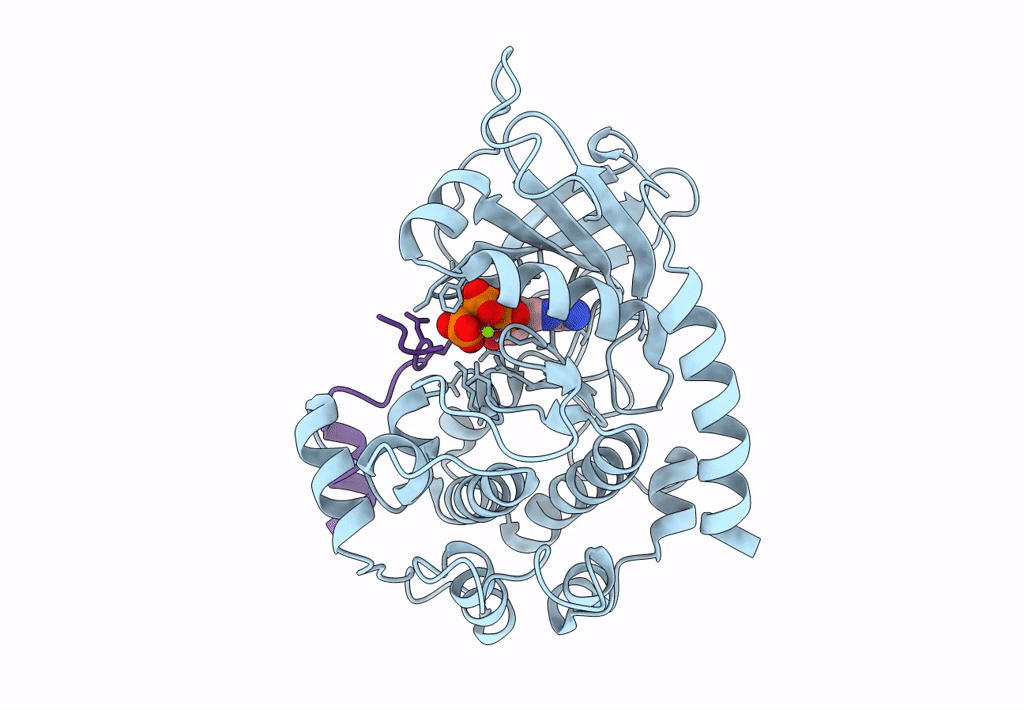
PDB ID:
1Q24
Keywords:
Title:
PKA double mutant model of PKB in complex with MgATP
Biological Source:
Source Organism(s):
Bos taurus (Taxon ID: 9913)
Expression System(s):
Method Details:
Experimental Method:
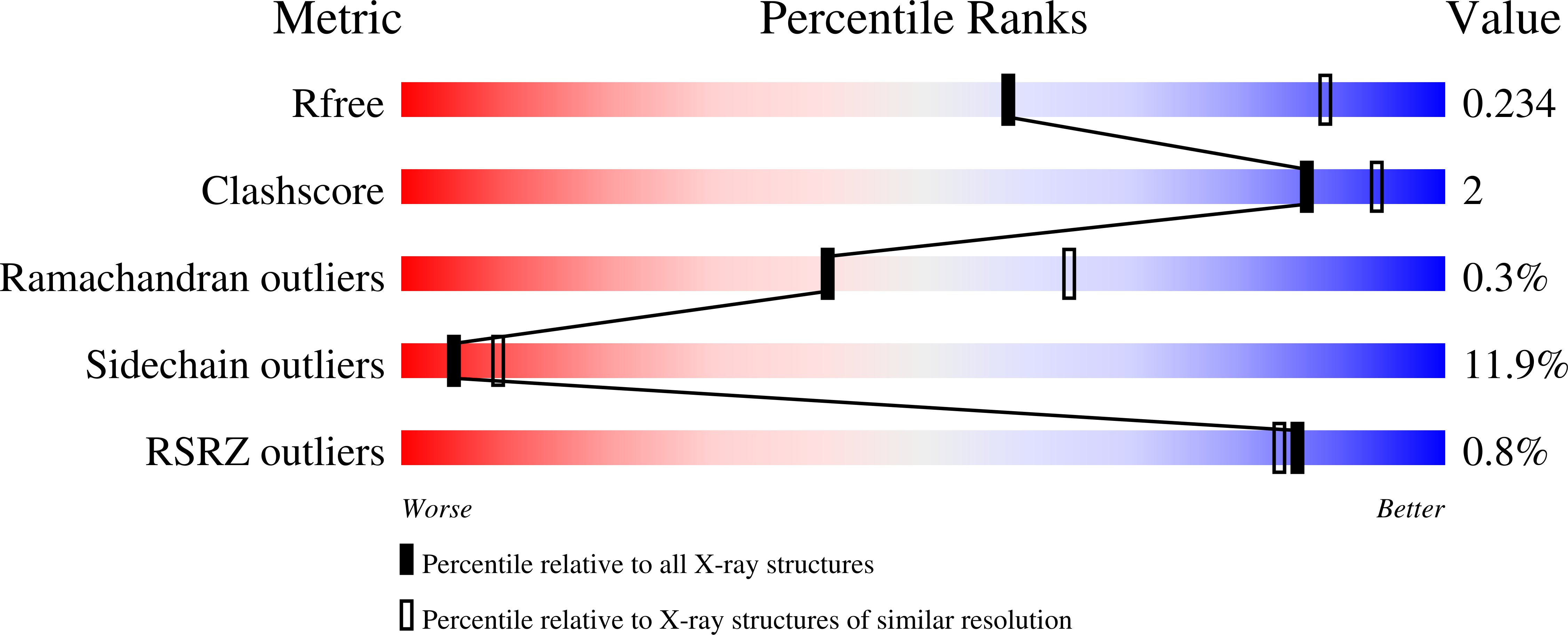
Resolution:
2.60 Å
R-Value Free:
0.25
R-Value Work:
0.19
R-Value Observed:
0.20
Space Group:
P 21 21 21