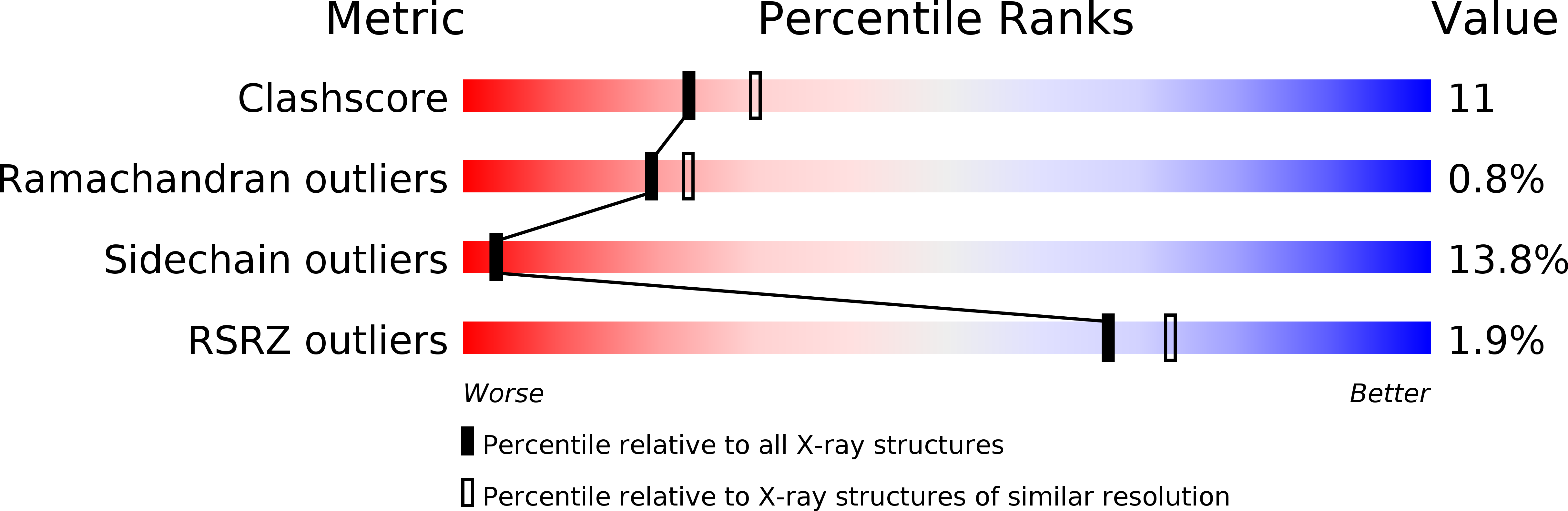
Deposition Date
1995-04-20
Release Date
1995-07-31
Last Version Date
2024-02-21
Entry Detail
PDB ID:
1PVD
Keywords:
Title:
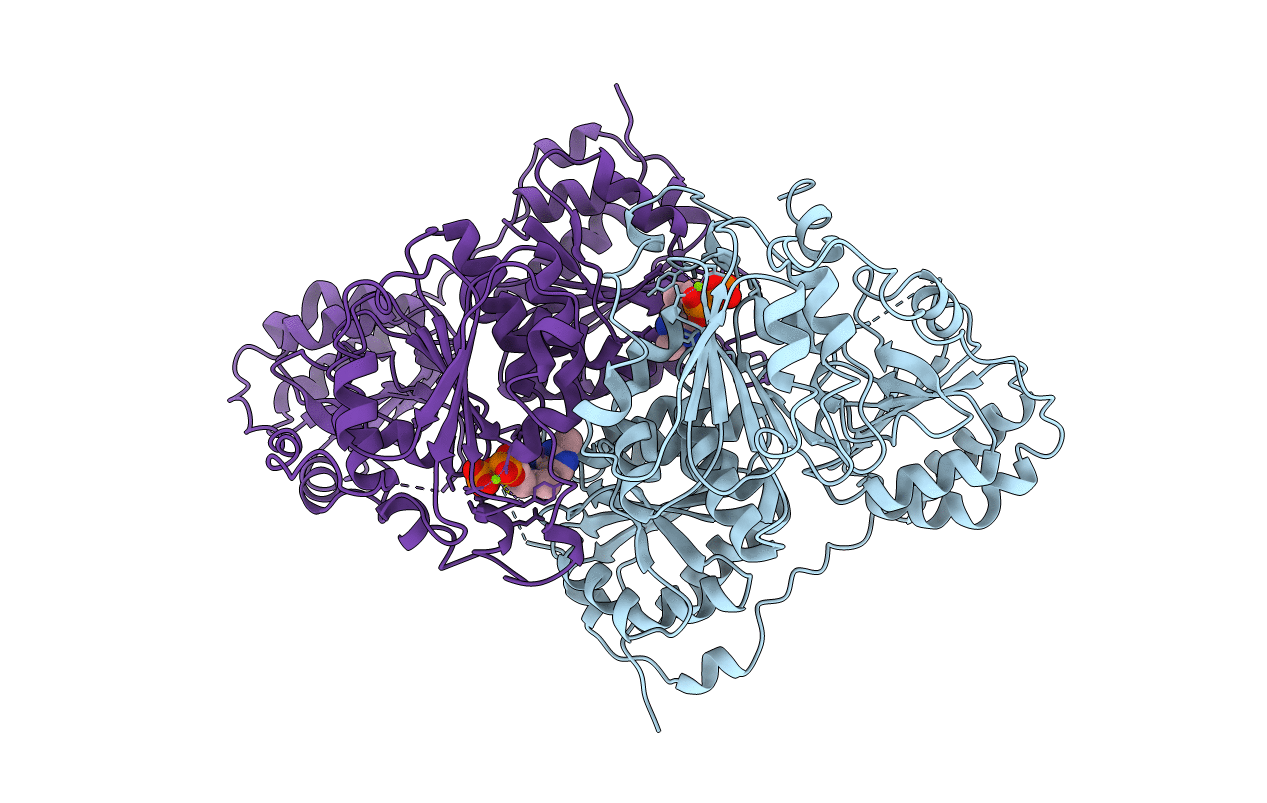
CRYSTAL STRUCTURE OF THE THIAMIN DIPHOSPHATE DEPENDENT ENZYME PYRUVATE DECARBOXYLASE FROM THE YEAST SACCHAROMYCES CEREVISIAE AT 2.3 ANGSTROMS RESOLUTION
Biological Source:
Source Organism(s):
Saccharomyces cerevisiae (Taxon ID: 4932)
Method Details:
Experimental Method:
Resolution:
2.30 Å
R-Value Work:
0.16
R-Value Observed:
0.16
Space Group:
C 1 2 1