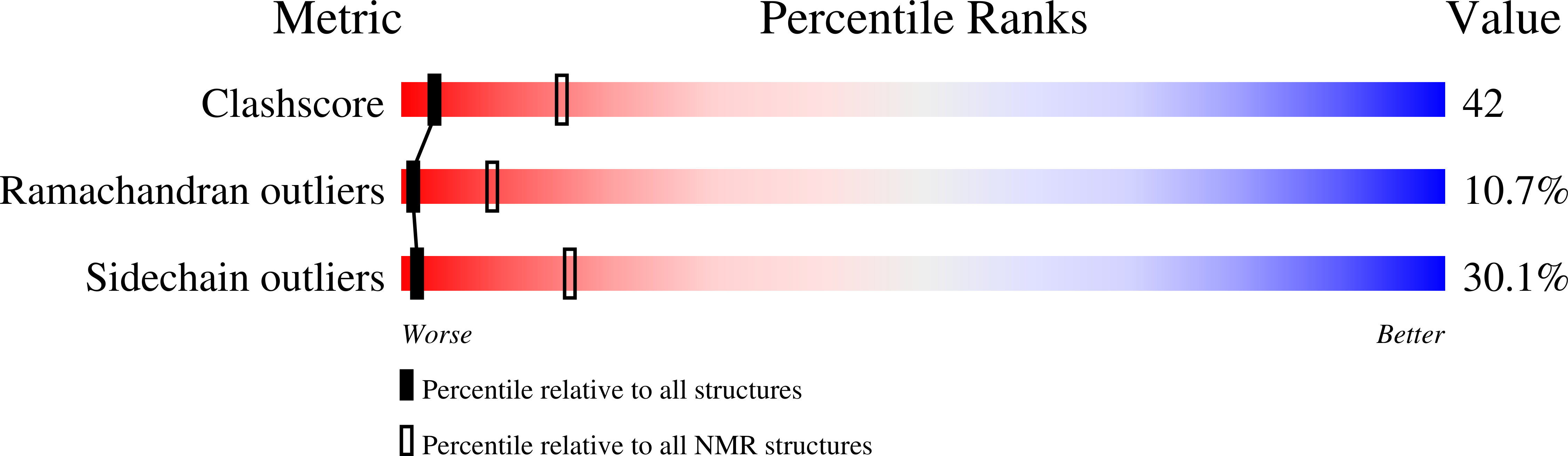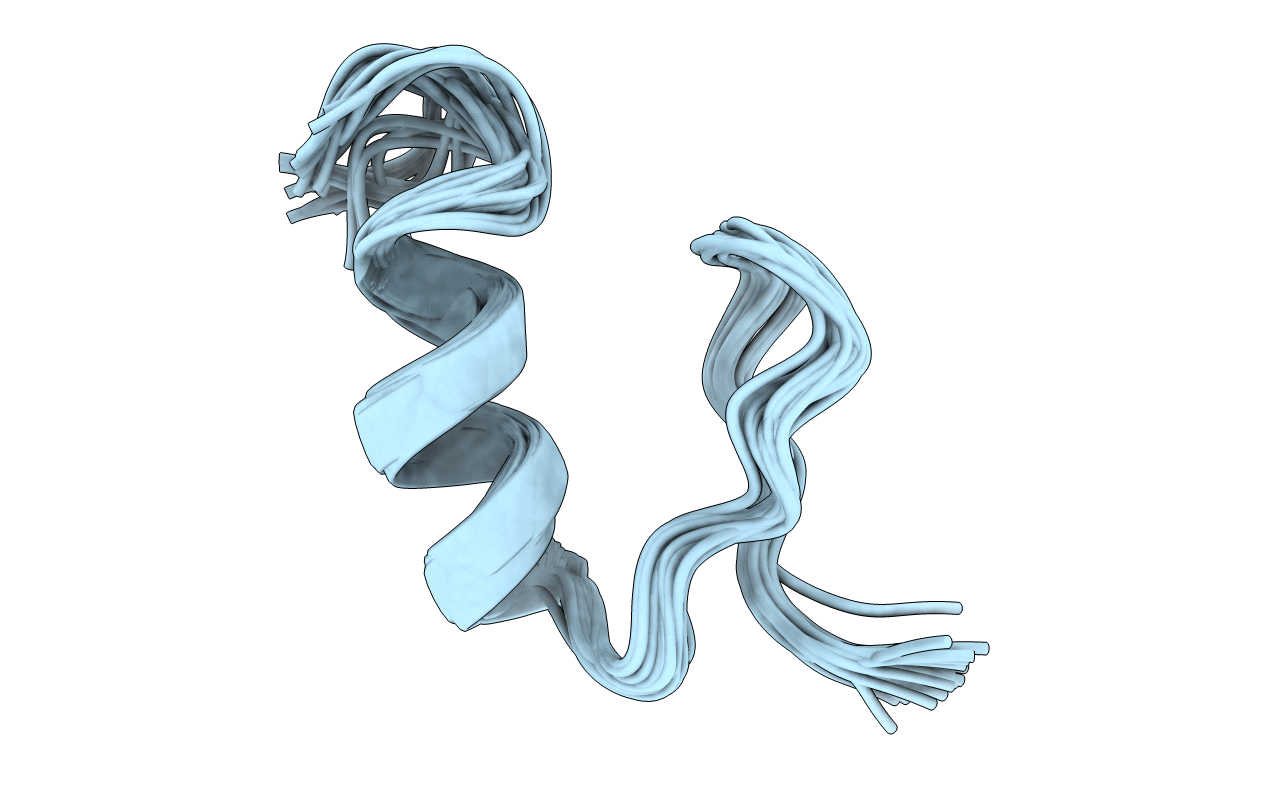
Deposition Date
1997-10-29
Release Date
1998-01-28
Last Version Date
2024-05-22
Entry Detail
PDB ID:
1PSV
Keywords:
Title:
COMPUTATIONALLY DESIGNED PEPTIDE WITH A BETA-BETA-ALPHA FOLD SELECTION, NMR, 32 STRUCTURES
Method Details:
Experimental Method:
Conformers Calculated:
98
Conformers Submitted:
32
Selection Criteria:
NO RESTRAINT VIOLATIONS GREATER THAN 0.3 ANGSTROMS, RMS DEVIATIONS FROM IDEALIZED BOND LENGTHS < 0.01 A, AND RMS DEVIATIONS FROM IDEALIZED ANGLES AND IMPROPERS < 1.0 DEGREE