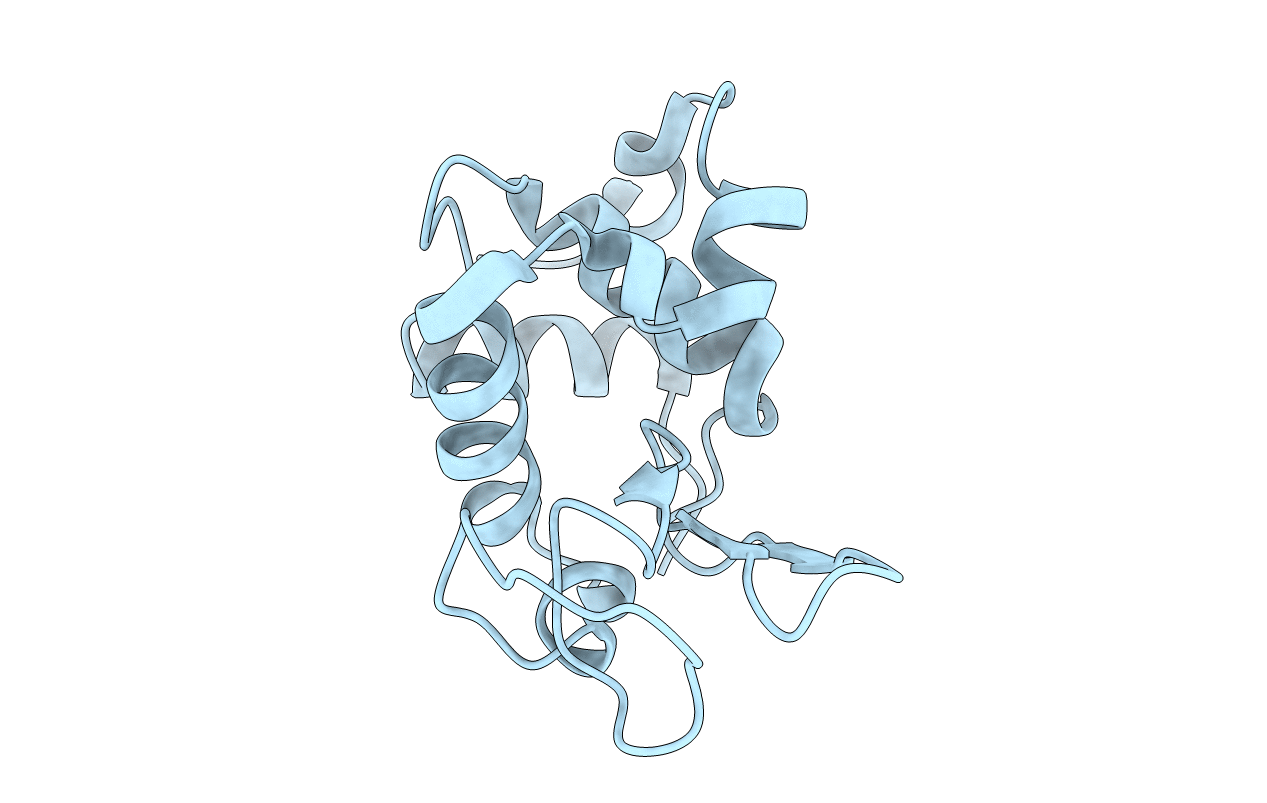
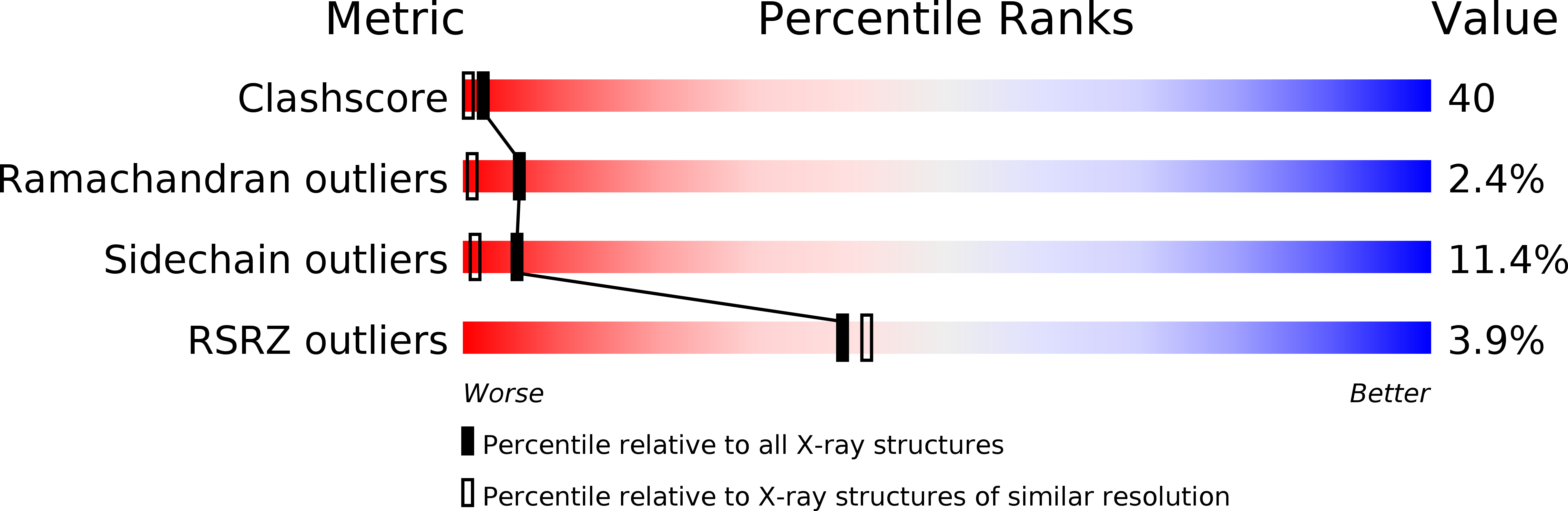X-ray diffraction data to 1.5 A resolution have been collected for triclinic crystals of hen egg white lysozyme. The triclinic model was derived from the tetragonal one by the rotation function and refined initially by Fo-Fc and differential difference syntheses against 2 A resolution data. Refinement was continued by differential difference cycles against the 1.5 A data until R was reduced to 0.220. Although the initial refinement was rapid, it was subsequently a matter of attrition, leading to a complete recheck of the data and the discovery of systematic error which affected primarily the high-resolution data. Refinement was continued against the corrected 2 A data by block-diagonal least squares. After five cycles the refinement was terminated at R = 0.254 because of the imminent availability of a preferred refinement program. Problems with the protein model, the solvent, and the interaction of the scale and thermal parameters are discussed. The experiences gained in this study are summarized.
Legend
Protein
Chemical
Disease