Deposition Date
2001-11-30
Release Date
2003-06-03
Last Version Date
2023-08-16
Entry Detail
PDB ID:
1KHT
Keywords:
Title:
Adenylate kinase from Methanococcus voltae
Biological Source:
Source Organism(s):
Methanococcus voltae (Taxon ID: 2188)
Expression System(s):
Method Details:
Experimental Method:
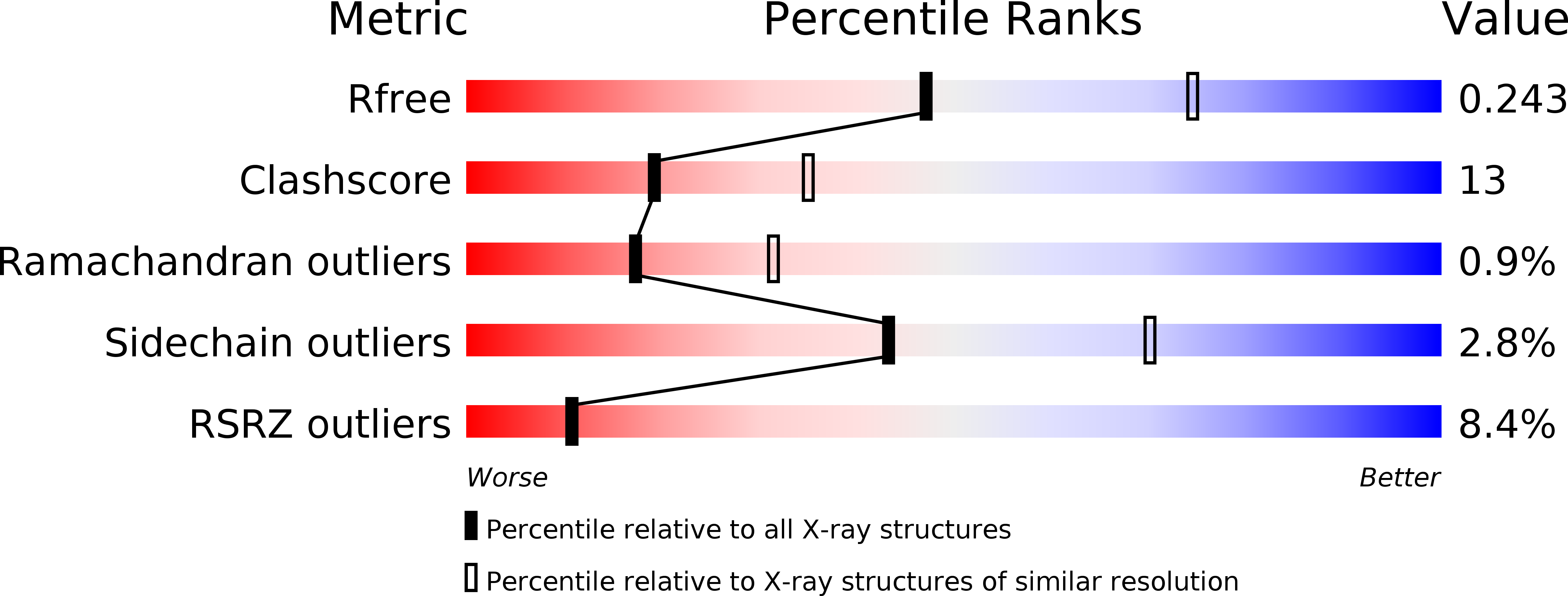
Resolution:
2.50 Å
R-Value Free:
0.25
R-Value Work:
0.22
R-Value Observed:
0.22
Space Group:
I 2 3