Deposition Date
2001-11-12
Release Date
2001-11-28
Last Version Date
2024-11-06
Entry Detail
PDB ID:
1KDD
Keywords:
Title:
X-ray structure of the coiled coil GCN4 ACID BASE HETERODIMER ACID-d12La16I BASE-d12La16L
Method Details:
Experimental Method:
Resolution:
2.14 Å
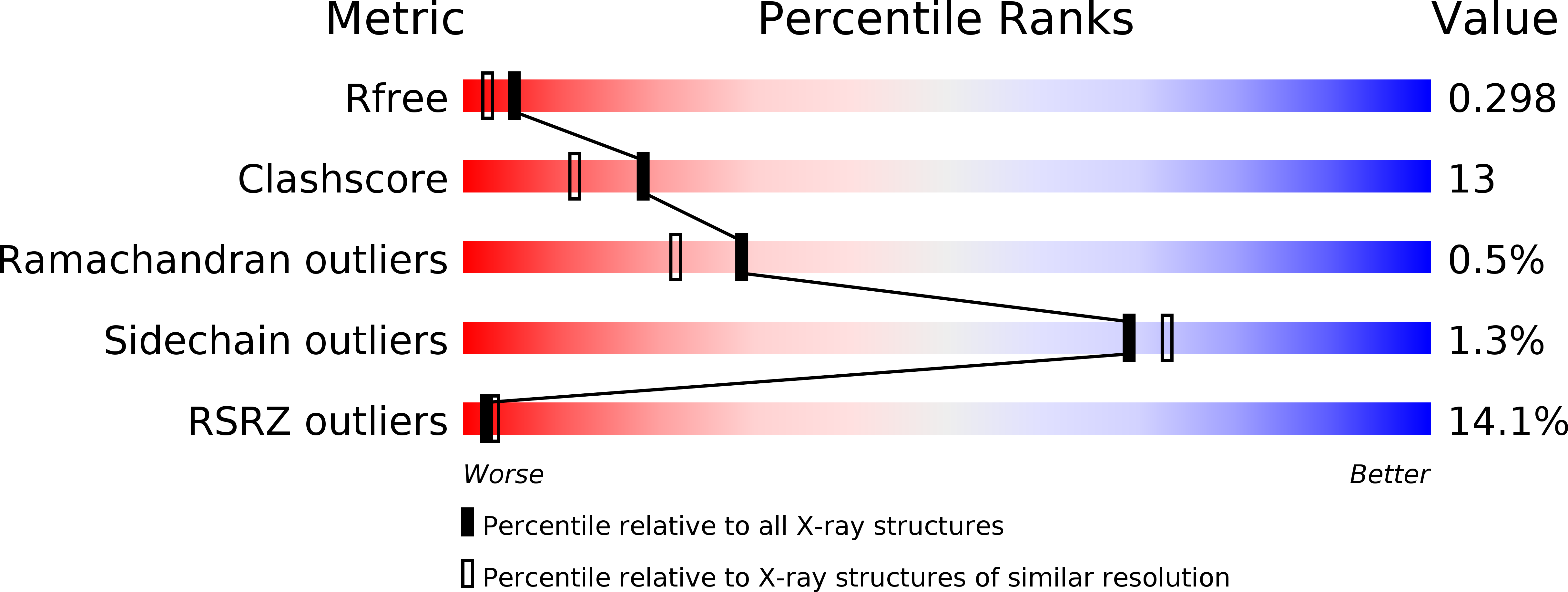
R-Value Free:
0.30
R-Value Work:
0.25
Space Group:
P 41 21 2