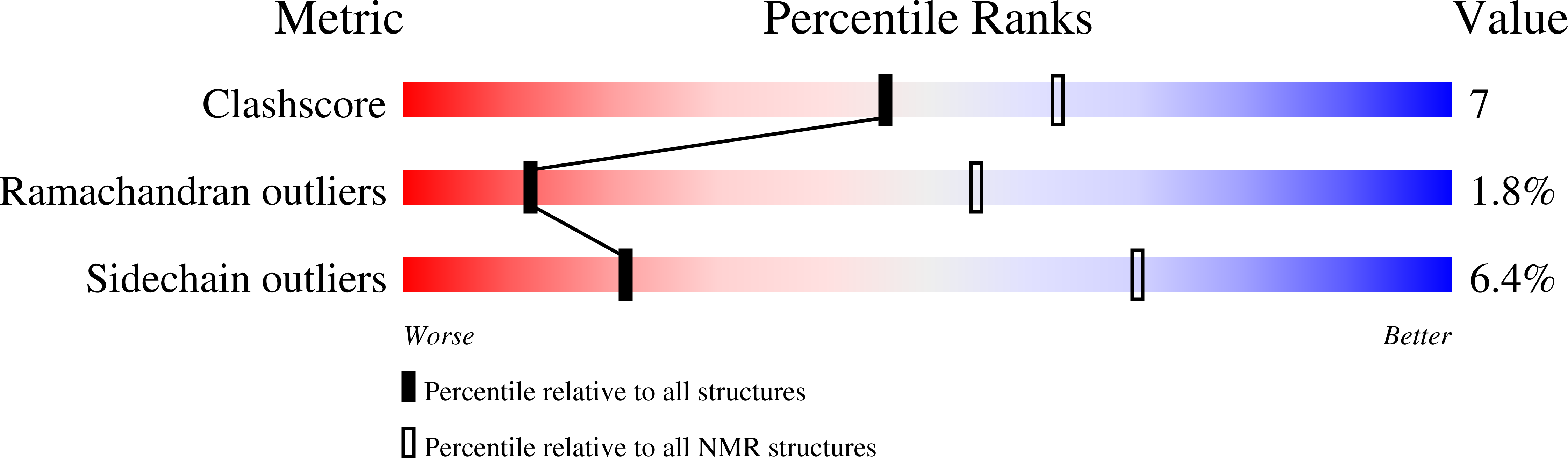
Deposition Date
2001-01-02
Release Date
2001-07-18
Last Version Date
2024-11-13
Entry Detail
PDB ID:
1HTX
Keywords:
Title:
SOLUTION STRUCTURE OF THE MAIN ALPHA-AMYLASE INHIBITOR FROM AMARANTH SEEDS
Biological Source:
Source Organism(s):
Amaranthus hypochondriacus (Taxon ID: 28502)
Method Details:
Experimental Method:
Conformers Calculated:
100
Conformers Submitted:
20
Selection Criteria:
structures with favorable non-bond energy,structures with the least restraint violations