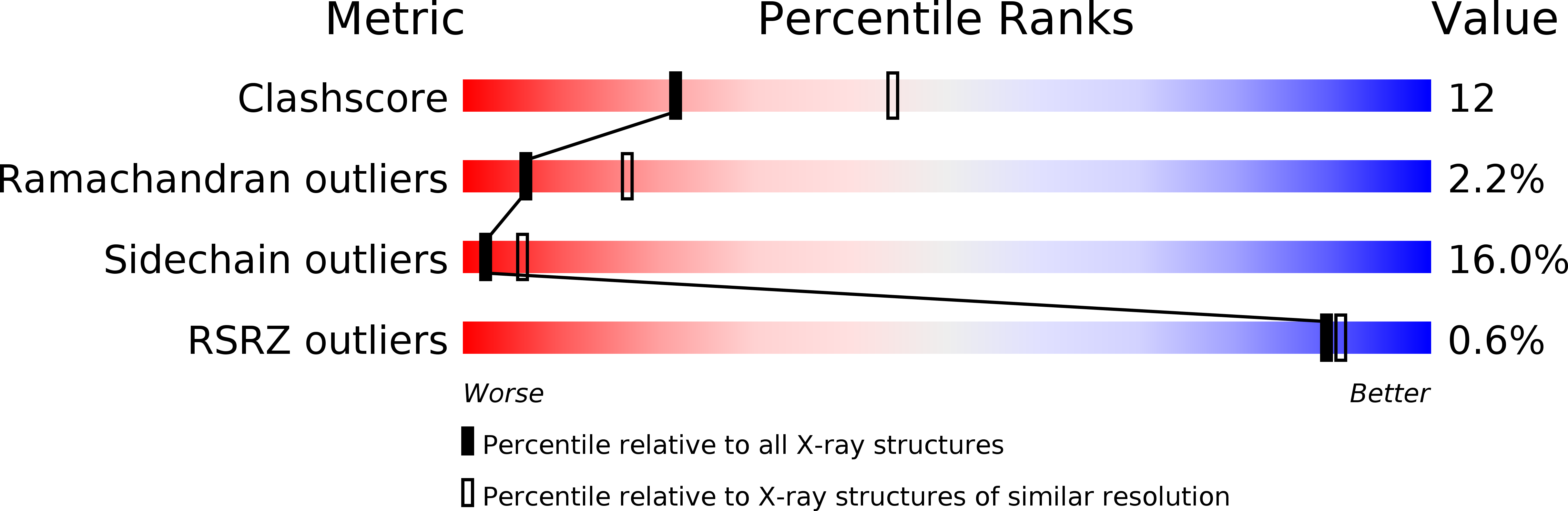
Deposition Date
1992-10-20
Release Date
1993-10-31
Last Version Date
2024-10-16
Entry Detail
PDB ID:
1FVE
Keywords:
Title:
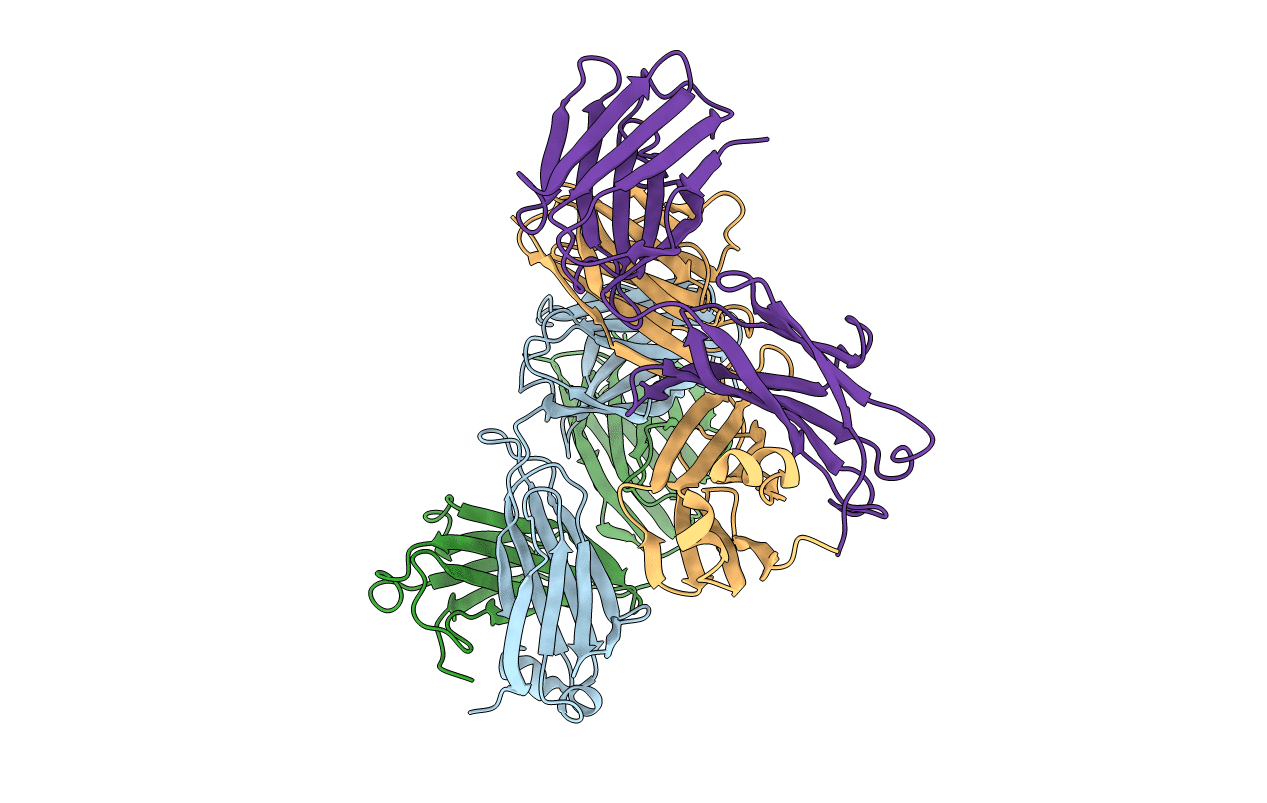
X-RAY STRUCTURES OF THE ANTIGEN-BINDING DOMAINS FROM THREE VARIANTS OF HUMANIZED ANTI-P185-HER2 ANTIBODY 4D5 AND COMPARISON WITH MOLECULAR MODELING
Biological Source:
Source Organism(s):
Homo sapiens (Taxon ID: 9606)
Expression System(s):
Method Details:
Experimental Method:
Resolution:
2.70 Å
R-Value Work:
0.17
R-Value Observed:
0.17
Space Group:
P 1