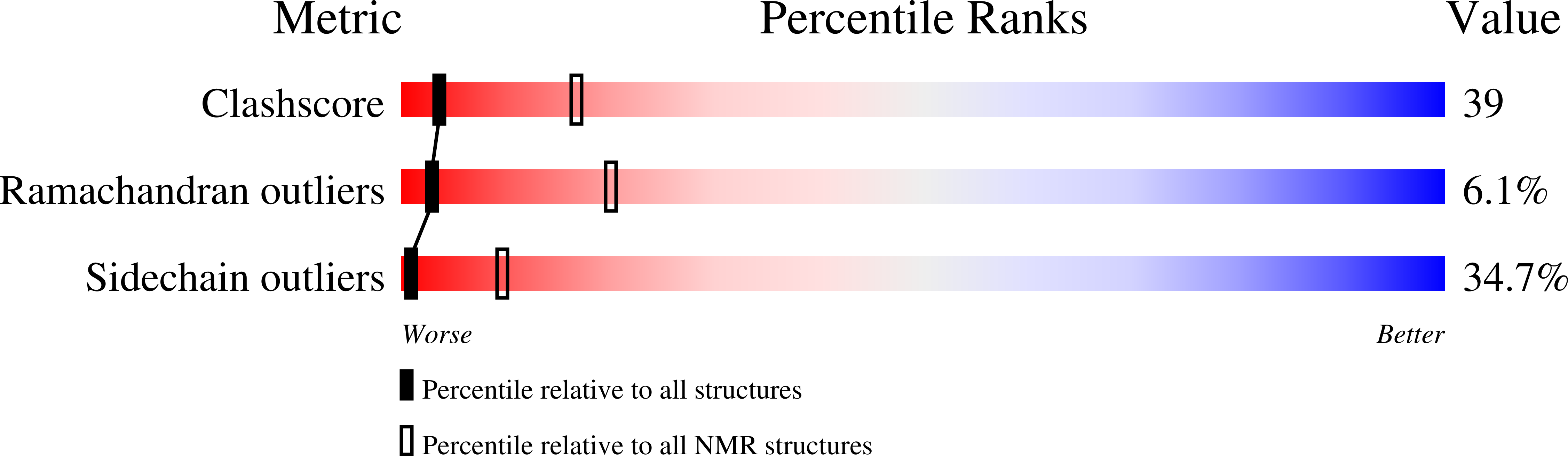
Deposition Date
2000-05-11
Release Date
2001-05-08
Last Version Date
2024-05-22
Entry Detail
PDB ID:
1EZP
Keywords:
Title:
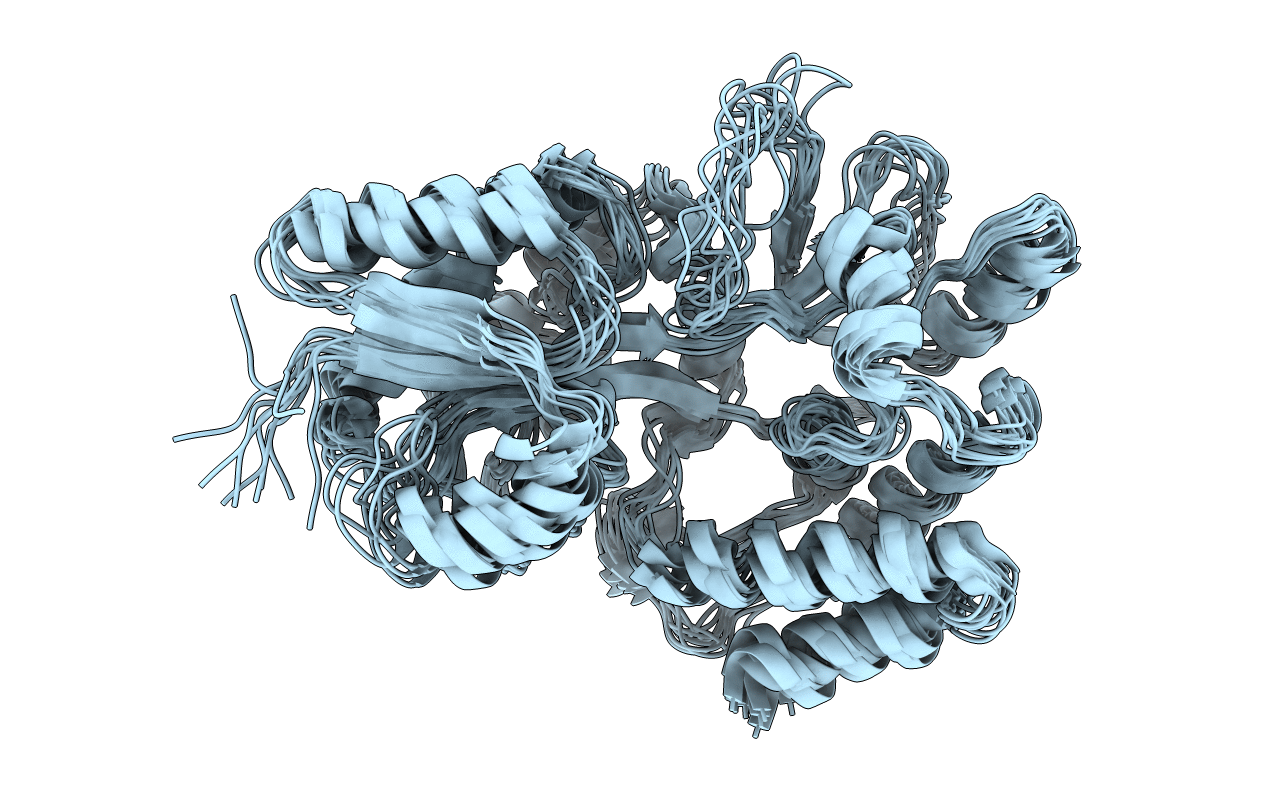
GLOBAL FOLD OF MALTODEXTRIN BINDING PROTEIN COMPLEXED WITH BETA-CYCLODEXTRIN USING PEPTIDE ORIENTATIONS FROM DIPOLAR COUPLINGS
Biological Source:
Source Organism(s):
Escherichia coli (Taxon ID: 562)
Expression System(s):
Method Details:
Experimental Method:
Conformers Calculated:
243
Conformers Submitted:
10
Selection Criteria:
structures with the lowest energy