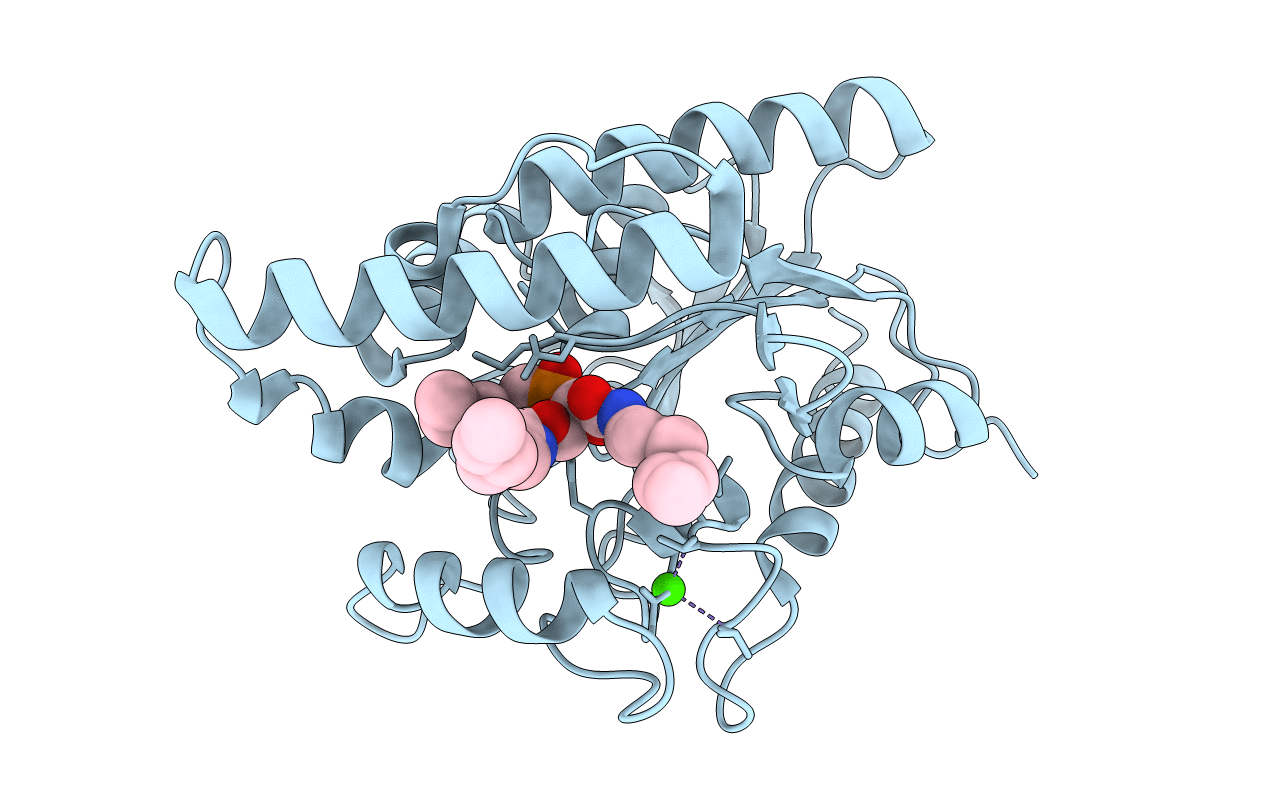
Deposition Date
2000-05-02
Release Date
2000-10-18
Last Version Date
2024-10-09
Entry Detail
PDB ID:
1EX9
Keywords:
Title:
CRYSTAL STRUCTURE OF THE PSEUDOMONAS AERUGINOSA LIPASE COMPLEXED WITH RC-(RP,SP)-1,2-DIOCTYLCARBAMOYL-GLYCERO-3-O-OCTYLPHOSPHONATE
Biological Source:
Source Organism(s):
Pseudomonas aeruginosa (Taxon ID: 287)
Expression System(s):
Method Details:
Experimental Method:
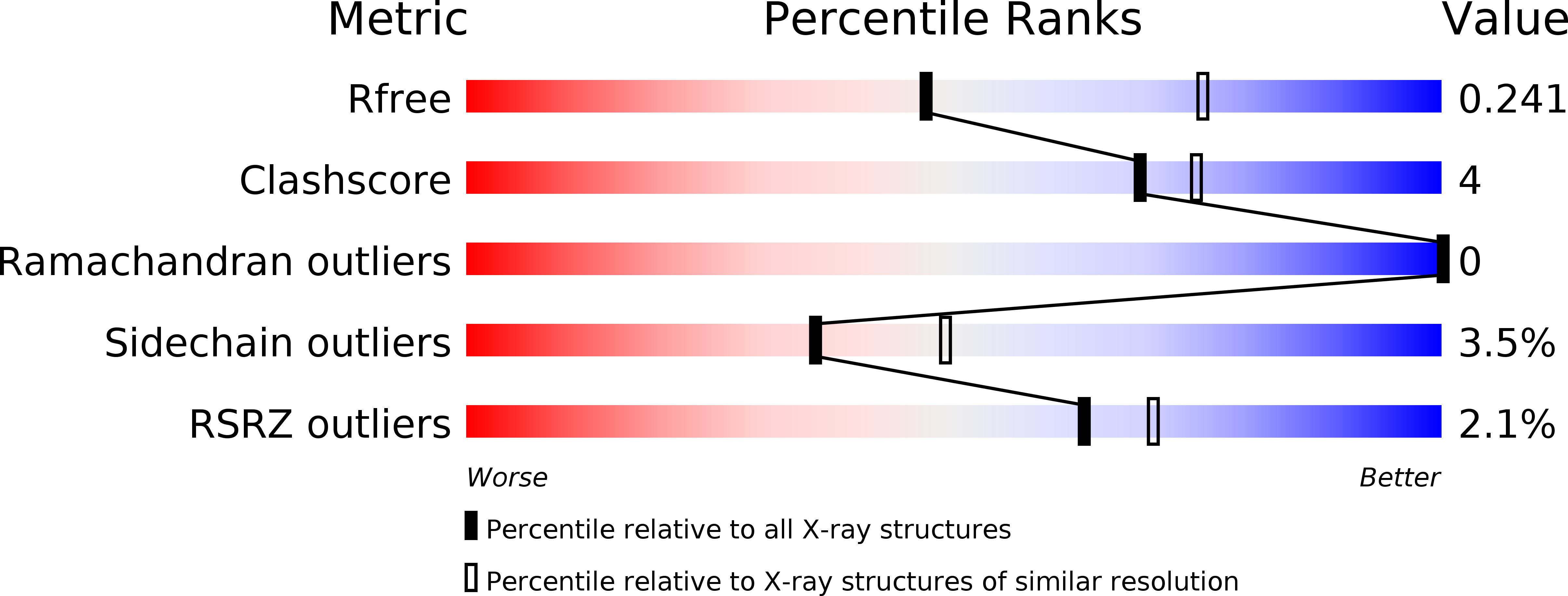
Resolution:
2.54 Å
R-Value Free:
0.24
R-Value Work:
0.19
R-Value Observed:
0.19
Space Group:
P 21 21 21