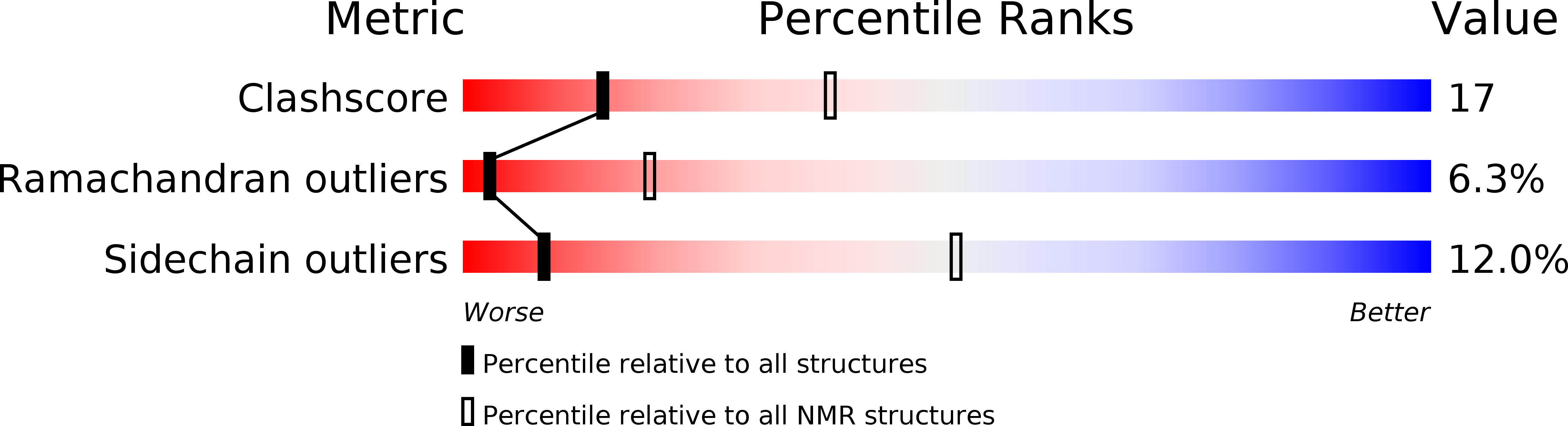Deposition Date
1999-12-08
Release Date
1999-12-10
Last Version Date
2024-11-20
Entry Detail
PDB ID:
1DWL
Keywords:
Title:
The Ferredoxin-Cytochrome complex using heteronuclear NMR and docking simulation
Biological Source:
Source Organism(s):
DESULFOMICROBIUM NORVEGICUM (Taxon ID: 52561)
DESULFOVIBRIO VULGARIS (Taxon ID: 882)
DESULFOVIBRIO VULGARIS (Taxon ID: 882)
Expression System(s):
Method Details:
Experimental Method:
Conformers Submitted:
3
Selection Criteria:
COMBINATION OF NMR SHIFT VARIATION AND A SOFT DOCKING ALGORITHM