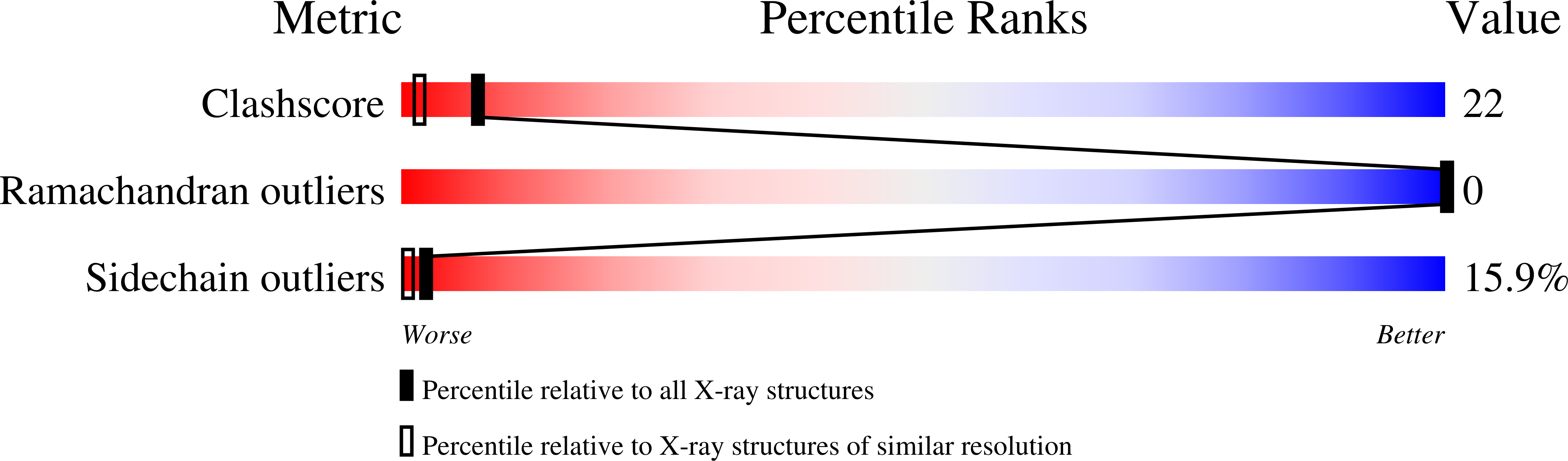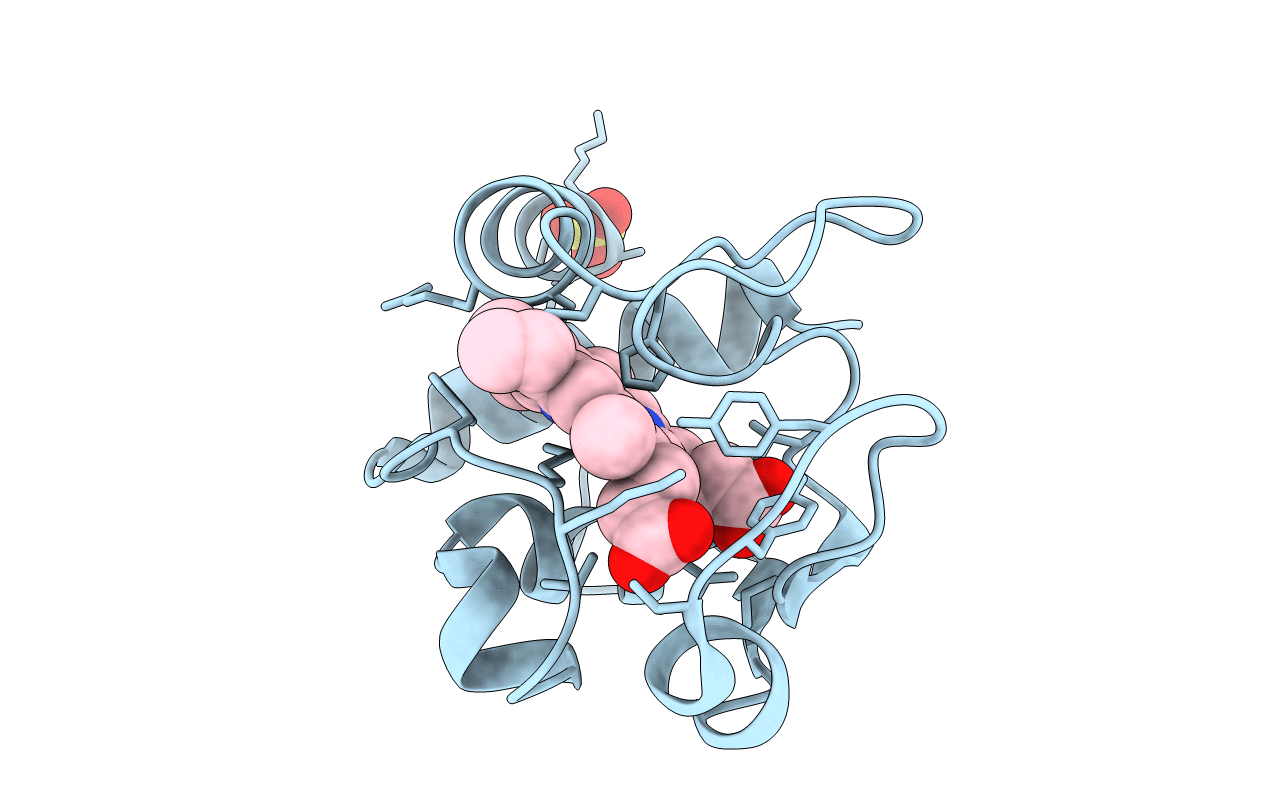
Deposition Date
1994-06-01
Release Date
1994-12-20
Last Version Date
2025-03-26
Entry Detail
PDB ID:
1CHH
Keywords:
Title:
STRUCTURAL STUDIES OF THE ROLES OF RESIDUES 82 AND 85 AT THE INTERACTIVE FACE OF CYTOCHROME C
Biological Source:
Source Organism:
Saccharomyces cerevisiae (Taxon ID: 4932)
Method Details:
Experimental Method:
Resolution:
1.97 Å
R-Value Observed:
0.18
Space Group:
P 43 21 2