
Deposition Date
1998-04-27
Release Date
1999-03-23
Last Version Date
2024-10-30
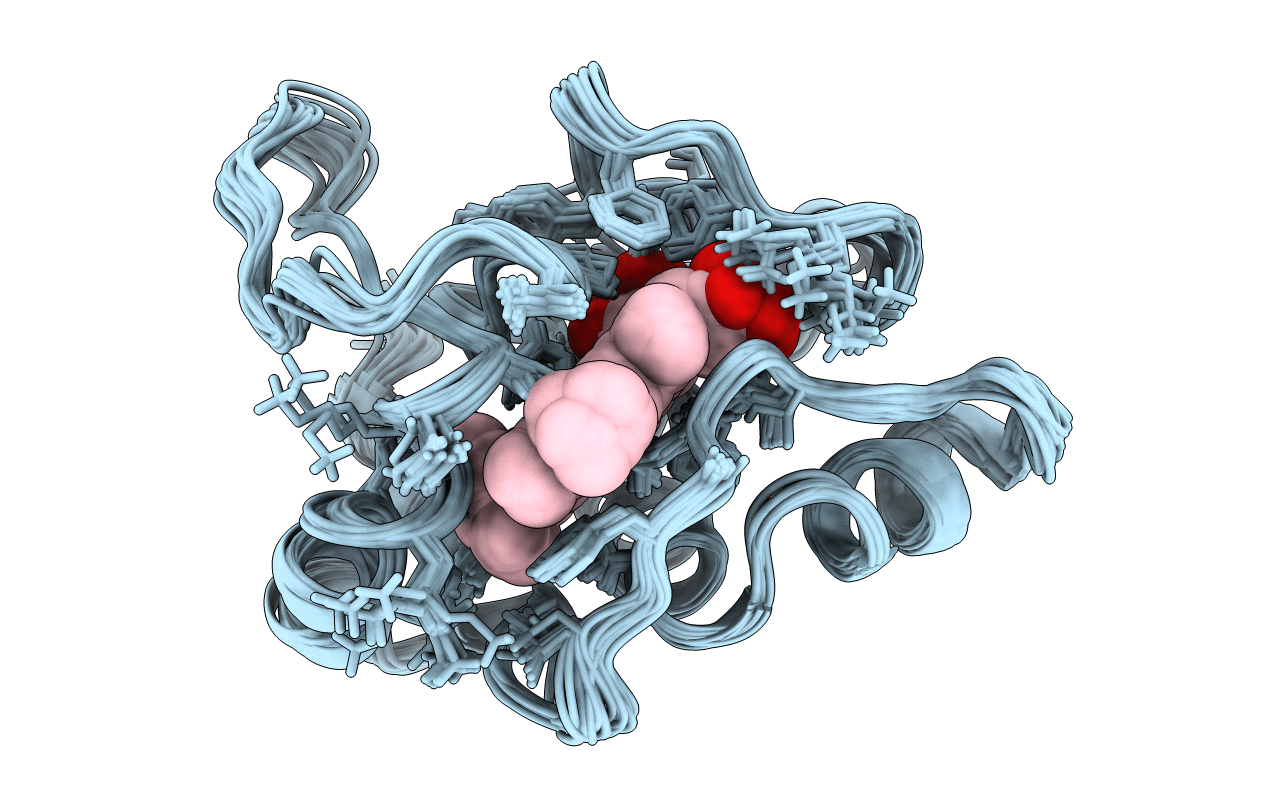
Method Details:
Experimental Method:
Conformers Calculated:
25
Conformers Submitted:
20
Selection Criteria:
LEAST RESTRAINT VIOLATION
